海带褐藻糖胶的分离纯化及其抗氧化活性测定
2022-02-16李荣蓉付燕红赵紫涵付学军孙利芹
李荣蓉,付燕红,赵紫涵,阮 新,付学军,孙利芹
(1.烟台大学 生命科学学院,山东 烟台 264005;2.烟台海关技术中心,山东 烟台 264000)
海带(Laminaria japonica.)中含有褐藻胶、褐藻糖胶、褐藻淀粉[1]等多糖。目前,我国海带的工业利用率仅占30%[2],其深加工主要集中在碘、褐藻胶及甘露醇等的生产中[3],造成海带价格瓶颈,制约了海带产业的发展,同时海带工业残渣作为废弃物,其中还有丰富的蛋白质、褐藻糖胶及膳食纤维,造成资源浪费。
褐藻糖胶(Fucoidan)是一种水溶性硫酸杂多糖,主要成分为硫酸化的α-L-岩藻糖,并含少量木糖、甘露糖、半乳糖和糖醛酸等[4]。近年来,大量研究已证明褐藻糖胶具有降血糖血脂、抗氧化、抗菌、抗肿瘤、调节机体免疫力等生理活性作用,已成为海洋药物研究的热点,有很好的应用前景[5-12]。目前,国内外对褐藻糖胶的研究较集中于提取率的提高,在进一步分离纯化和生理活性研究上不够深入。褐藻糖胶的初步分离纯化通常采用乙醇分级沉淀法、CaCl2法和季胺盐沉淀法等方法[13]。但其提取物中通常会含有蛋白质、色素、褐藻胶等杂质[14],采用Sevag法、三氯乙酸法、鞣酸法[15-16]等方法去除蛋白会损失褐藻糖胶,同时会影响褐藻糖胶活性[17],因此需要对褐藻糖胶分离纯化方法、结构和抗氧化活性进行进一步研究。
采用DEAE-Sepharose Fast Flow弱阴离子交换柱层析及高效液相凝胶色谱(High performance liquidchromatography,HPLC)对海带中褐藻糖胶粗品进行分离纯化工艺的研究,测定了高效液相凝胶色谱分离所得3个组分的分子量,并对DEAE-Sepharose Fast Flow弱阴离子交换柱层析得到的3个组分清除DPPH(1,1-二苯基-2-三硝基苯肼)自由基和·OH(羟自由基)能力进行测定,分析不同分子量褐藻糖胶的抗氧化活性。研究为海带褐藻糖胶的高效且安全的分离纯化方法提供试验依据,有利于有效开发利用具有生物活性的海洋藻类多糖[18],减少资源浪费,提高海带的附加值,更好地开发和利用海洋资源[19]。
1 材料与方法
1.1 材料与试剂
干海带,山东荣成寻山集团有限公司提供;纤维素酶(20 000 U/g)、果胶酶(60 000 U/g)、复合蛋白酶(390 000 U/g),帝斯曼公司提供;中性蛋白酶(20 000 U/mL),无锡市酶制剂厂提供;标准葡聚糖,中国计量科学研究院提供;其余试剂均为分析纯。
1.2 仪器与设备
TU-1810型紫外可见分光光度计,北京普析通用仪器有限责任公司产品;Agilent1100型高效液相色谱仪,安捷伦科技有限公司产品;CBS-A型程控全自动部分收集器、DHL-A型电脑数显恒流泵,上海沪西分析仪器厂有限公司产品。
1.3 试验方法
1.3.1 褐藻糖胶粗品制备方法
工艺流程:干海带→粉碎、过40目筛→柠檬酸-褐藻酸钠缓冲液浸提→复合酶解→升温灭酶→NaOH溶液碱溶消化→过滤→滤液加HCl溶液酸凝过夜→加Na2CO3溶液中和→4倍体积乙醇醇析→减压抽滤→沉淀→干燥→褐藻糖胶粗品。
酶解条件[20]:纤维素酶添加量1%,中性蛋白酶添加量0.5%,复合蛋白酶添加量0.5%,果胶酶添加量1%,料液比1∶40,pH值6.0,温度60℃,时间3 h。
1.3.2 褐藻糖胶组分的分离
(1)DEAE-Sepharose FF弱阴离子交换柱层析分离褐藻糖胶[21]。分别称取不同质量(0.1,0.2,0.3,0.4 g)褐藻糖胶粗品,用0.02 mol/L的磷酸盐缓冲液(Phosphate-Buffered Saline,PBS,pH值7.4)配成5,10,15,20 mg/mL不同质量浓度褐藻糖胶溶液,分别上样于DEAE-Sepharose FF弱阴离子交换柱(1.6 cm×30 cm),用0~2.0 mol/L的NaCl溶液(pH值7.4,0.02 mol/L的PBS配制)线性梯度洗脱,洗脱流速0.5 mL/min,自动部分收集器15 min/管收集。苯酚-硫酸法[22]测定多糖含量,以管数为横坐标,吸光度为纵坐标,绘制相应的洗脱曲线,收集各洗脱峰,超滤浓缩后冷冻干燥。
(2)高效液相凝胶色谱分离褐藻糖胶。取适量褐藻糖胶粗品,用超纯水配制成200 μg/mL的样品溶液,过0.45 μm微孔滤膜,4℃下冷藏,备用。
色谱条件:Agilent1100型高效液相色谱系统;Waters UltrahydrogelTM linear色谱柱(7.8 mm×300 mm),示差折光(RI)检测器,流动相:超纯水,流速0.6 mL/min,柱温40℃,进样量20 μL。
(3)HPLC测定褐藻糖胶组分相对分子质量。采用1.3.2高效液相色谱条件,将HPLC分离得到的褐藻糖胶样品组分和一系列不同相对分子质量的标准葡聚糖(12.6,60.6,110,521 kD)分别流经Agilent1100型高效液相色谱系统中Waters UltrahydrogelTM linear色谱柱,标准品质量浓度为200 μg/mL,示差折光检测器检测。由标准葡聚糖的相对分子质量对数与保留时间做标准曲线,以此计算样品相对分子质量及分布,样品纯度由峰型判断。
1.3.3 体外抗氧化活性测定
(1)清除DPPH自由基的能力。将褐藻糖胶样品组分(D-1,D-2,D-3)配成质量浓度分别为0.2,0.4,0.6,0.8,1.0 mg/mL,各取2 mL于试管中,分别加入0.1 mmol/L的DPPH乙醇溶液2 mL,混匀静置20 min后测定波长517 nm处的光吸度(用乙醇2 mL+水2 mL作参比)。同时,以相同浓度梯度的BHT(2,6-二叔丁基-4-甲基苯酚)作为对照,重复上述操作,并设置平行。
DPPH自由基清除能力的计算公式:

式中:Ai——DPPH溶液2 mL+样品溶液2 mL的吸光度;
Aj——乙醇2 mL+样品溶液2 mL的吸光度;
Ac——乙醇2 mL+DPPH溶液2 mL的吸光度。
(2)清除·OH的能力。根据Fenton反应原理测定褐藻糖胶清除·OH的能力。将褐藻糖胶(D-1,D-2,D-3)配制成质量浓度为0.1,0.2,0.4,0.6,0.8,1.0,1.2,1.4,1.6 mg/mL。取6 mmol/L水杨酸1 mL于试管中,加入2 mmol/L FeSO4溶液1 mL混匀,再加入6 mmol/L H2O2溶液1 mL混匀,于37℃下恒温水浴15 min,于波长510 nm处测定其吸光度A损伤(用水作为参比)。测定后,各加入上述质量浓度的海带多糖溶液1 mL,混匀静置10 min,再测定其吸光度A加样,并用水代替H2O2的A未损伤做参照。同时,以相同质量浓度的维C作为对照,重复上述操作,并设置平行。
·OH清除能力的计算公式:

式中:A损伤——反应体系原来的吸光度;
A未损伤——加入提取液但不加入H2O2的吸光度;A加样——反应体系加入提取液后的吸光度。
2 结果与分析
2.1 DEAE-Sepharose FF弱阴离子交换色谱分离褐藻糖胶
对5,10,15,20 mg/mL不同质量浓度褐藻糖胶溶液,NaCl溶液0~2.0 mol/L梯度洗脱时的分离效果。
褐藻糖胶不同上样量的离子交换色谱梯度洗脱曲线见图1。
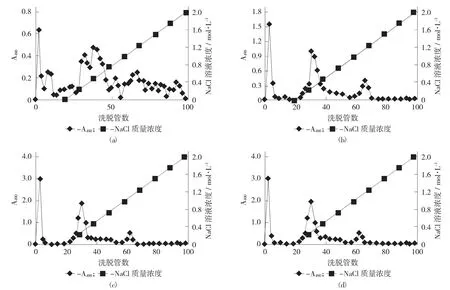
图1 褐藻糖胶不同上样量的离子交换色谱梯度洗脱曲线
由图1可知,图(a)~(d)分别是上样质量浓度为5,10,15,20 mg/mL的洗脱图。当上样质量浓度为5 mg/mL时,各组分分离不彻底,且含量较少,不便于收集(见图1(a));由图1(c)和图1(d),随着上样量增大,盐洗脱峰的多糖含量并没有增加,表明图1(c)上样质量浓度为15 mg/mL时,此时各组分分离程度比较好,且呈现单一对称峰,说明达到一定的纯度。因此,选择上样质量浓度15 mg/mL,既达到了良好的分离效果,又便于收集。
根据图1(c),试验共分离得到3个峰,分别记为D-1,D-2和D-3。其中,D-1为流出峰,D-2和D-3为盐洗脱峰,D-2 NaCl溶液浓度为0-0.5 mol/L,D-3 NaCl溶液浓度为1.00~1.25 mol/L。各峰型均无拖尾现象,分离效果较理想。
2.2 高效液相凝胶色谱分离褐藻糖胶
褐藻糖胶的HPLC色谱图见图2,HPLC色谱峰的保留时间和相对峰面积见表1。

表1 HPLC色谱峰的保留时间和相对峰面积
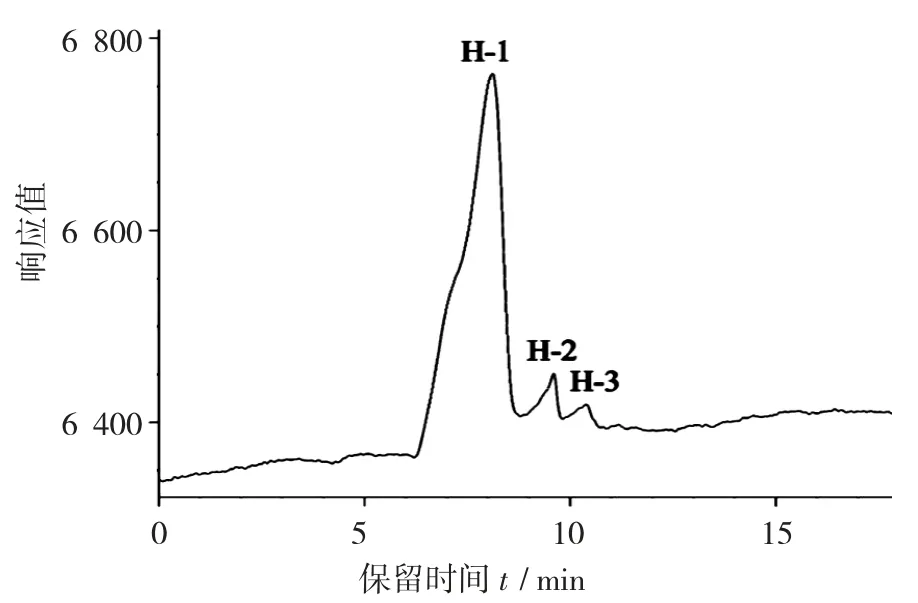
图2 褐藻糖胶的HPLC色谱图
褐藻糖胶样品经过高效液相凝胶色谱分出了3个组分H-1,H-2,H-3。其中,H-1的保留时间为8.107 min,H-2的保留时间为9.594 min,H-3的保留时间为10.397 min。
2.3 纯度与相对分子量测定结果
2.3.1 褐藻糖胶组分纯度
由图2可知,褐藻糖胶样品所分出的3个组分,高效液相色谱图均呈现单一窄峰,因此试验分离得到的3个组分可被认为是均一组分。
2.3.2 HPLC相对分子量测定结果
标准葡聚糖的HPLC色谱图见图3。
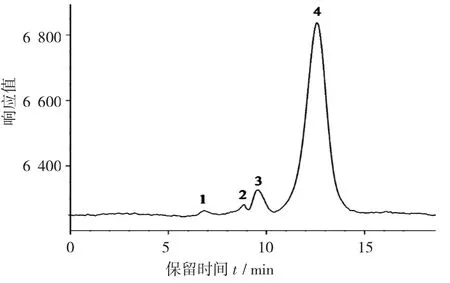
图3 标准葡聚糖的HPLC色谱图
根据标准葡聚糖的HPLC色谱图,各标准葡聚糖的出峰时间分别为6.824,8.852,9.576,12.573 min,与其对应的分子量为521,110,60.6,12.6 kD。
标准葡聚糖标准工作曲线见图4。

图4 标准葡聚糖标准工作曲线
根据图4葡聚糖标准工作曲线和图2褐藻糖胶样品中各组分的出峰时间可得3个组分的分子量大小。
色谱峰的保留时间和分子量见表2。

表2 色谱峰的保留时间和分子量
2.4 褐藻糖胶清除自由基的测定结果
2.4.1 褐藻糖胶清除DPPH自由基的测定结果
褐藻糖胶各组分和BHT的DPPH自由基清除见图5。
由图5可知,在试验质量浓度范围内,褐藻糖胶3个组分D-1,D-2,D-3和BHT对DPPH自由基都有一定的清除作用,随着质量浓度的增加,DPPH自由基的清除率也在增大,均呈一定剂量-效应相关性,清除率大小为BHT>H-3>H-2>H-1。测定BHT和褐藻糖胶各组分的IC50值,以BHT抗氧化剂作为对照,不同分子量褐藻糖胶组分清除DPPH自由基能力均低于BHT,BHT的IC50为0.922 8 mg/mL。褐藻糖胶中D-3的IC50最小,为1.254 0 mg/mL,表示对DPPH自由基的清除能力最强;其次是D-2,IC50为1.874 0 mg/mL;D-1对DPPH自由基的IC50最大,为2.412 2 mg/mL,对DPPH自由基的清除能力最弱。3个组分由强到弱排列顺序为D-3>D-2>D-1,对比分子量发现,褐藻糖胶的分子量越小,对DPPH自由基的清除能力越强,体外抗氧化效果越好。
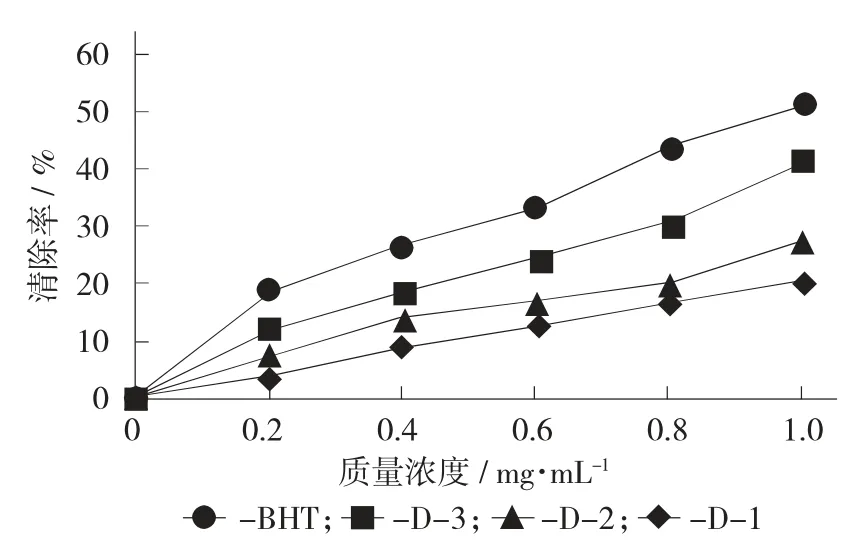
图5 褐藻糖胶各组分和BHT的DPPH自由基清除率
2.4.2 褐藻糖胶清除羟基自由基的测定结果
褐藻糖胶各组分和维C的·OH清除率见图6。

图6 褐藻糖胶各组分和维C的·OH清除率
由图6可知,在试验质量浓度范围内,褐藻糖胶3个组分D-1,D-2,D-3和维C对·OH都有一定的清除作用,随着质量浓度的增加,·OH的清除率也在增大,呈一定剂量-效应相关性,当升高到一定质量浓度时,清除率基本趋于定值。测定维C和褐藻糖胶各组分的IC50值,以维C作为对照,维C的IC50为0.349 8 mg/mL,褐藻糖胶组分中D-3的IC50小于维C,D-1和D-2的IC50大于维C。褐藻糖胶D-3的IC50最小,为0.306 5 mg/mL,表示对·OH的清除能力最强;其次是D-2,IC50为0.549 1 mg/mL;D-1的IC50最大,为0.795 1 mg/mL,对·OH的清除能力最弱。由此可见,褐藻糖胶对·OH有较强的清除能力,3个组分由强到弱排列顺序为D-3>D-2>D-1,即褐藻糖胶的分子量越小,对·OH的清除能力越强,体外抗氧化活性越好。
3 讨论
利用高效液相凝胶色谱对海带褐藻糖胶粗品进行分离纯化,达到很好的分离效果,分离纯化的同时可测出各组分的分子量。该分离方法相较于离子交换层析和凝胶柱层析等其他分离方法,具有样品处理简单、灵敏度高、制备方便快速、样品用量少且不被破坏等优点,已有相关文献报道高效液相色谱在褐藻糖胶研究中用于测定组分的单糖组成[23-24],但采用高效凝胶色谱分离褐藻糖胶组分并对各组分进行分子量的测定相关文献少见。
对高效液凝胶相色谱纯化得到的褐藻糖胶3个组分进行了分子量测定,结果为193.148,74.407,44.452 kD。与周军明[25]用离子交换层析纯化褐藻糖胶后采用Sephadex G-150型凝胶柱层析测的各组分分子量为120 226.4 D和13 182.5 D的结果有一定差异,可能因多糖种类不同导致。用高效液凝胶相色谱测定多糖分子量要比Sephadex G-150型凝胶柱层析更为快速、精确。试验还测定了DEAE-Sepharose FF弱阴离子交换柱层析纯化得到的褐藻糖胶3个组分清除DPPH自由基和·OH的能力与分子量的相关性,与陶传云等人[26]研究褐藻糖胶多糖的体外抗氧化性能结果相一致,均证明了分子量越小的组分,体外抗氧化效果越好。由于时间限制,没有进一步研究D-1,D-2,D-3 3个组分与H-1,H-2,H-3 3个组分的对应关系及相关性,褐藻糖胶各组分的组成、结构及生物体内的活性还需要继续深入研究和探索。
4 结论
(1)采用2种方法对海带褐藻糖胶粗品进行分离纯化,结果表明,DEAE-Sepharose FF弱阴离子交换柱层析和高效液相凝胶色谱均分离出3个组分,适于褐藻糖胶的纯化[27]。
(2)DEAE-Sepharose FF弱阴离子交换柱层析的最佳条件为褐藻糖胶粗品上样量为20 mL(质量浓度为15 mg/mL),0.5 mL/min,0~2.0 mol/L的Na-Cl溶液进行线性梯度洗脱,得到的3个峰分别为D-1,D-2,D-3,其中D-1为流出峰,D-2和D-3为盐洗脱峰。高效液相凝胶色谱分离所得H-1,H-2,H-3共3个组分,分子量分别为193.148,74.407,44.452 kD。
(3)D-1,D-2,D-3组分对DPPH自由基和·OH自由基均表现出一定的清除能力,3个组分清除DPPH自由基能力均低于BHT;D-3清除·OH的能力高于维C,D-1和D-2清除·OH的能力低于维C。且3个组分对2种自由基的清除率随质量浓度升高而增大,清除2种自由基能力由强到弱的排列顺序均为D-3>D-2>D-1,即分子量越小,体外抗氧化活性越高。
