磷酸二酯酶的促肿瘤作用及其抑制剂抗肿瘤作用研究进展
2022-02-15张作艳李杨玲朱素燕
张作艳,李杨玲,周 旋,林 珠,朱素燕,徐 萍,张 翀
(1.宁波市第一医院药学部,浙江 宁波 315000;2.浙江大学医学院附属杭州市第一人民医院临床药学部,浙江 杭州 310006;3.浙大城市学院医学院,浙江 杭州 310015)
磷酸二酯酶(phosphodiesterase,PDE)是一类可以特异性水解环磷酸腺苷(cyclic adenosine monophosphate,cAMP)和环磷酸鸟苷(cyclic guanosine monophosphate,cGMP)的酶类,而 cAMP/cGMP表达水平与能量代谢、记忆、免疫、细胞增殖、周期调节和凋亡等密切相关[1]。PDE家族成员众多,分布于人体的各个组织和器官,且不同亚型在不同组织器官中的表达不尽相同。PDE在中枢神经系统、免疫、血液和泌尿系统疾病的发生发展发挥重要作用,因其广泛的的抑制效果,PDE抑制剂的研究开发成为近几年研究的热点。
PDE家族由11个亚型组成,包括PDE1~PDE11,这些PDE具有降解cAMP、cGMP和调节细胞功能的能力[1]。部分PDE可特异性水解cAMP(PDE4,PDE7和PDE8),一些PDE则水解cGMP(PDE5,PDE6和PDE9),而有些PDE则能同时水解两者(PDE1,PDE2,PDE3,PDE10 和 PDE11)[2-3]。近年来,陆续有研究发现,PDE可通过调节细胞周期和诱导细胞凋亡等机制参与肿瘤进展,另外PDE在肿瘤组织中的异常表达也可作为生物标志物[4-5]。研究发现,相较于人类正常组织或其他癌组织,PDE3A和PDE3B在胃肠道间质瘤(gastrointestinal stromal tumors,GIST)中表达较高,可作为GIST的生物标志物[6]。此外,PDE4A在肝癌患者组织中表达显著高于癌旁组织,且PDE4A表达较高肝癌患者肝切除术后预后也较差,PDE4A在肝癌中诱导上皮间质转化[7],促进肿瘤转移,可作为肿瘤预测因子及肝癌治疗靶点。本文就PDE的结构与功能及其基因亚型和抑制剂在肿瘤中的研究进展进行综述,并对PDE参与调控肿瘤的分子机制进行分析,为研究开发特异性强,生物活性高的PDE抑制剂和调控肿瘤发生的分子机制及临床应用提供思路。
1 磷酸二酯酶的结构与功能
1.1 结构
PDE包括N端调节结构域和C端催化结构域。催化结构域高度保守,包含cAMP和cGMP分子底物结合口袋、同源结构域和氨基酸残基等,是金属依赖性磷酸水解酶的独特特征结构域,保证PDE各亚型调节特性及细胞功能[5];不同亚型调节结构域氨基酸序列差别很大,如图1所示,主要有Ca2+/钙调蛋白(calmodulin,CaM)结合位点(PDE1),cGMP磷酸二酯酶-腺苷酸环化酶-细菌转录因子1A(cGMP-binding phosphodiesterases,adenylyl cyclases,formate hydrogenlyase tracription activator 1A,GAF)催化结构域(PDE2,5,6,10,11),膜相关结构域(PDE3),上游保护区(upstream conserved regions,UCR)结构域(PDE4)和周期昼夜蛋白-芳基烃受体核转运蛋白-专一蛋白(Per-Arnt-Sim,PAS)结构域(PDE8),以及蛋白激酶A(protein kinase A,PKA)和cGMP依赖性蛋白激酶(cGMP-dependent protein kinase,PKG)磷酸化的必要位点如丝氨酸157(Ser157),丝氨酸239(Ser239)等[1,8-9]。
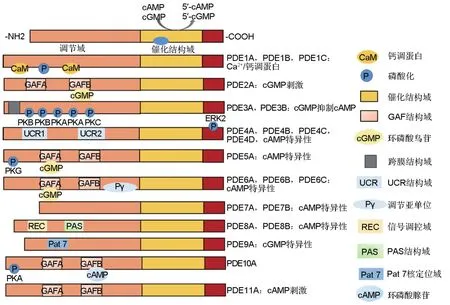
图1 磷酸二酯酶(PDE)各亚型结构与功能.GAF:cGMP磷酸二酯酶-腺苷酸环化酶-细菌转录因子;PK:蛋白激酶;PKG:cGMP依赖性蛋白激酶.
1.2 功能
cAMP和cGMP作为第二信使,通过激活PKA/PKG通路参与人体多种生理作用。PDE作为水解cAMP和cGMP的调节因子,通过调控PDE表达影响人体生理病理过程。PDE通过mRNA剪切或转录产生近百种同工酶[10],各亚型的三维结构、动力学特性、调节模式、细胞内定位、细胞表达和抑制剂的敏感性均不同[11],但是通过cGMP/cAMP信号通路(抑制肿瘤细胞生长,是PDEs发挥促肿瘤作用的主要途径。研究表明[12],PDE1可抑制人恶性黑色素瘤相关抗原细胞人白血病细胞增殖。研究发现,PDE1作为分化诱导因子(differentiation inducing factors-1,DIF-1)的特异性靶标,DIF-1 0.5 mmol·L-1可竞争性阻断cAMP与PDE1结合,发挥肿瘤抑制作用[12]。PDE3B在结肠癌细胞HT-29中高表达,研究发现环状磷脂酸(cyclic phosphatidic acid,cPA)可抑制 PKB磷酸化,而PKB磷酸化后,以浓度依赖性方式激活PDE3B,下调cAMP表达水平,促进HT-29细胞增殖[13],推测cPA/PDE3B/cAMP信号通路是抑制结肠癌细胞增殖的主要信号通路。
2 磷酸二脂酶抑制剂的抗癌作用及机制
PDE作为一个多基因家族,可通过cAMP水解细胞内的调节其生物学功能。研究发现,多个PDE抑制剂在抗肿瘤中发挥较大作用,能够抑制肿瘤细胞生长。如,PDE1抑制剂能够通过诱导肝癌细胞自噬增加对索拉非尼(sorafenib)的敏感性,促进肝癌细胞凋亡[14];PDE4抑制剂在临床试验中对肝癌和胶质母细胞瘤具有较好的抑制效果。PDE1~PDE11各亚型组成、常见抑制剂以及调节结构域和组织分布表达见表1。
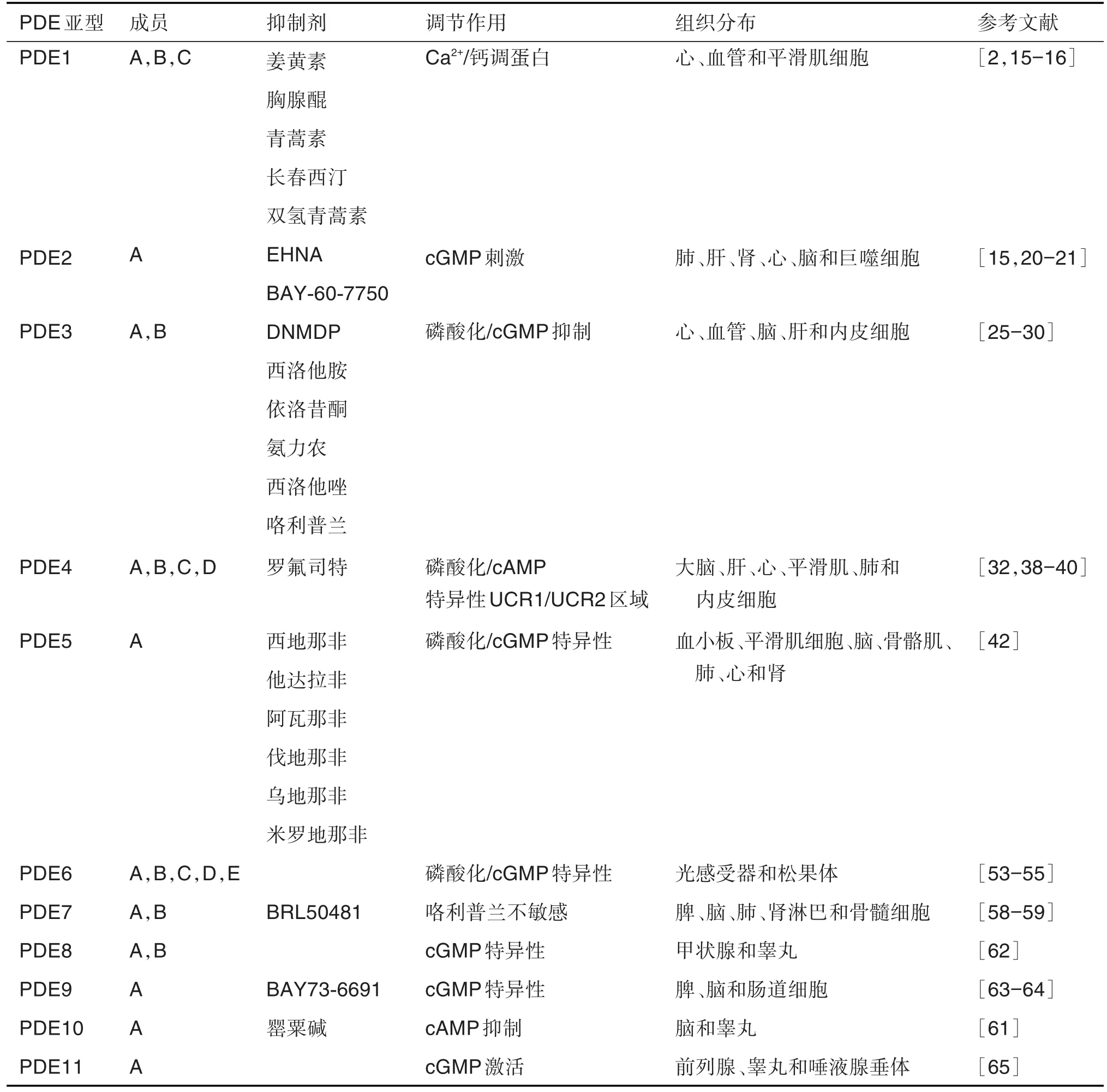
表1 磷酸二酯酶(PDE)各亚型成员、抑制剂及组织分布
2.1 磷酸二酯酶1抑制剂
PDE1是最早发现的同工酶,能够同时水解cGMP和cAMP,有A,B,C 3种亚型。PDE1发挥作用主要是通过Ca2+结合到PDE1的CaM结构域,Ca2+和CaM绑定后,缓解催化位点N端自动抑制,从而发挥作用。PDE1抑制剂主要有长春西汀、姜黄素、尼莫地平、胸腺醌、青蒿素和双氢青蒿素等。胸腺醌可特异性抑制急性淋巴白血病细胞PDE1A表达,通过激活P73抑制jurkat细胞生长和结直肠癌细胞增殖[15]。姜黄素抑制PDE1后,细胞周期蛋白激酶表达增强,抑制肿瘤细胞增殖[16]。研究发现,靶向CaM抑制PDE1活性是PDE1抑制剂发挥抗肿瘤作用的重要机制,如天然倍半萜内脂(tehranolide)和柚皮素通过靶向CaM使PDE1A酶活丧失,细胞内cAMP增加,激活PKA,最终抑制白血病细胞增殖[17-18]。
2.2 磷酸二酯酶2抑制剂
PDE2可同时水解cGMP和cAMP,PDE2结合cGMP后,可促进cAMP水解,因此又称为cGMP驱动型PDE2。PDE2仅有PDE2A一种亚型,目前已知的PDE2抑制剂有外消旋-9-(2-羟基-3-壬基)腺嘌呤盐酸盐〔racemic-9-(2-hydroxy-3-nonyl)adenine9 hydrochloride,EHNA〕,Bay60-7750,IC933和双嘧达默(dipyridamole)等。研究发现,EHNA可通过调节细胞周期发挥抗肿瘤作用[19-20],如EHNA通过抑制黑色素瘤细胞DNA合成,使肿瘤细胞停滞在G2/M期,周期蛋白A mRNA水平下降影响黑色素瘤细胞生长[15]。此外,PDE2在人骨肉瘤组织中高表达,抑制PDE2-cAMP途径可抑制骨肉瘤细胞增殖,而抑制PDE2-cGMP途径抑制骨肉瘤细胞转移[21]。线粒体稳态在细胞生长中发挥关键作用,研究发现,线粒体Ca2+可激活结肠癌细胞中PDE2活性,通过cAMP/PKA信号通路上调线粒体转录因子,促进结肠癌细胞生长[22]。PDE2A主要分布在线粒体内,可通过PKA介导线粒体接触部位和嵴组织系统磷酸化,调节Parkin向线粒体募集和诱导线粒体自噬[23]。
2.3 磷酸二酯酶3抑制剂
PDE3主要包括PDE3A和PDE3B。PDE3主要表达在心、血管、大脑、肝和内皮细胞[24]。研究发现,PDE3A在不同实体瘤患者的肿瘤细胞亚群中均有表达,但以GIST标本表达最高。92%GIST患者肿瘤组织中检测到PDE3A高表达[25],且患者PDE3A表达水平越高,抑制剂化疗效果也更明显,因此,PDE3A有望成为肿瘤生物标志物,用于判断癌症治疗效果[26-27]。西洛酰胺(cilostamide)和西洛他唑(cilostazol)是PDE3特异性抑制剂,西洛他唑目前已获FDA批准,用于治疗间歇性跛行、肺动脉高压和慢性阻塞性肺疾病。研究发现,西洛他唑和西洛酰胺可有效抑制肿瘤细胞增殖和侵袭[28],还能减弱GIST对伊马替尼耐药[29]。此外,Kumazoe等[30]发现,PDE3抑制剂和表没食子儿茶素没食子酸酯联用可抑制肿瘤干细胞因子FOXO3和CD44在胰腺癌细胞中表达,抑制胰腺癌形成和肝转移。
2.4 磷酸二酯酶4抑制剂
PDE4包括4种亚型(PDE4A,PDE4B,PDE4C和PDE4D),约有20个同工酶,对cAMP高度特异,广泛分布在大脑、肾、心肌细胞、内皮细胞和免疫细胞中[31]。目前对PDE4的研究主要集中在免疫和炎症相关疾病。PDE4抑制剂发挥抗肿瘤作用主要通过诱导细胞凋亡、抑制肿瘤细胞迁移和诱导肿瘤活性细胞抑制因子生成。大多数情况下,选择性抑制PDE4活性可抑制癌细胞生长,如结直肠癌、胰腺癌、肝癌和急性淋巴白血病等,但却对黑色素瘤细胞有促生长作用[32-34]。PDE4A和PDE4D在肝癌细胞中异常高表达。另外,PDE4D还可用于评估前列腺癌患者预后,PDE4D表达低的患者术后风险增加[35]。Rho是肿瘤转移的临床诊断指标。研究发现,PDE4C抑制剂可上调RhoA和RhoC表达,可诱导RhoA磷酸化进而抑制胰腺癌转移[36]。由此可见,PDE4与肿瘤进展密切相关,可作为肿瘤生物标记物和治疗靶点。
第一代PDE4抑制剂主要有茶碱、咯利普兰(rolipram)和吡拉米司特(piclamilast)等。其中,咯利普兰对肝癌细胞和胶质母细胞瘤抑制作用显著,在临床试验中也具有良好疗效[37-38]。成胶质细胞瘤干细胞能分泌PDE4,促进血管内皮生长因子(vascular endothelial growth factor,VEGF)表达,咯利普兰和VEGF阻断剂贝伐单抗(bevacizumab)能有效诱导细胞凋亡,两种药物联用可抑制蛋白激酶B磷酸化[38]。VEGF与肿瘤转移密切相关,因此,靶向抑制PDE4对于胶质母细胞瘤转移也具有作用。
第二代PDE4抑制剂有罗氟司特(roflumilast)和西洛司特(cilomilast)等。罗氟司特使细胞内cAMP水平增加,可有效抑制成熟B细胞淋巴瘤细胞和白血病细胞增殖[39]。另外,一项Ⅰ期临床研究显示,罗氟司特和泼尼松用于治疗晚期B细胞肿瘤患者时,罗氟司特单药治疗显著抑制率患者磷脂酰肌醇3-激酶(phosphoinositide 3-kinase,PI3K)活性,且PI3K低表达患者存活的时间较PI3K高表达的患者时间增加了67 d,表明罗氟司特可用于临床B细胞恶性肿瘤的治疗[40]。
2.5 磷酸二酯酶5抑制剂
PDE5可特异性水解cGMP,仅有PDE5A一种亚型。PDE5A在多种肿瘤中过表达,如膀胱癌、前列腺癌、乳腺癌、结肠腺癌、慢性淋巴细胞白血病和非小细胞肺癌等[41],PDE5选择性抑制剂主要有西地那非(sildenafil)、伐地那非(vardenafil)、他达那非(tildenafil)和乌地那非(udenafil)等。PDE5抑制剂对肿瘤的抑制作用多与cGMP有关,其中涉及的信号分子有cGMP依赖性蛋白激酶(cGMP-dependent protein kinase,PKG)、腺苷酸激活蛋白激酶(AMP-activated protein kinase,AMPK)、哺乳动物Ste20样激酶/大肿瘤抑制激酶(large tumor suppressor/mammalian ste20-like kinase,MST/LATS)激酶和分子伴侣热休克蛋白90(human heat shock protein,HSP90)[42-43]。研究发现,抑制PDE5能够增强顺铂的敏感性,PDE5抑制剂与化疗药物联用能有效预防前列腺癌转移和复发[44]。西地那非作用于肿瘤细胞后,HSP90表达会增加,与HSP90抑制剂PU-H71联合用药可协同抑制体内癌细胞生长[45]。此外,PDE5在乳腺癌相关成纤维细胞中表达增加,且与患者总生存期相关,PDE5过表达会使胚胎成纤维细胞转化为活化的成纤维细胞表型,通过CXCL16促进乳腺癌细胞生长和转移[46]。
PDE5抑制剂可抑制结肠癌细胞生长,另外也可抑制手术诱导的免疫抑制,通过cGMP/PKG2信号转导抑制结直肠癌发展[47-48]。GUCY2C是一种鸟苷酸环化酶受体,GUCY2C结合鸟嘌呤后可激活cGMP下游信号通路,PDE5水解cGMP可阻断GUCY2C信号转导促进大肠癌进展[49]。一项临床调查显示,使用PDE5抑制剂能有效降低患大肠肿瘤的风险,且早期使用PDE5抑制剂的患者患病风险远小于晚期使用者[50],这种风险降低与PDE5抑制剂的累计剂量增加有关。另外,使用PDE5抑制剂可有效改善男性结直肠癌患者预后,降低转移风险[51]。
2.6 磷酸二酯酶6
PDE6又称为感光PDE,主要分布在视网膜中,是光感受器细胞进行光转换级联反应过程的重要酶[52]。PDE6每个亚基N端区域包含2个变构cGMP结合域GAF-A和GAF-B,两个结合域取向相同,可与C端的催化域相互作用发挥催化活性[53]。在黑色素瘤细胞中,PDE6可被癌症视网膜抗原通过Wnt5A-Frizzled-2激活,导致cGMP水平降低,CaM增加,钙稳态失衡[54],而钙稳态失衡会诱导活性转录因子cAMP通过调控微管网络调控原件(cAMP-response element binding protein,CREB)磷酸化,促进肿瘤进展。另外,在人乳腺癌细胞和患者原发性癌组织中检测到PDE6B,PDE6C和PDE6D的大量表达,说明PDE6在乳腺癌进展中可能发挥作用[55]。PDE6非选择性抑制剂扎普司特和双嘧达莫目前主要用于心血管和支气管哮喘等疾病的治疗,在肿瘤方面的作用尚未见报道。
2.7 磷酸二酯酶7抑制剂
PDE7生物学功能与PDE4相似,对cAMP有高度选择性,包括PDE7A和PDE7B 2种亚型,常见分布于大脑、脾、肺部、胸腺和淋巴细胞,尤以T淋巴细胞和巨噬细胞中表达最高。目前,PDE7对T淋巴细胞的作用存在争议,有些研究者发现PDE7可促进T细胞激活,也有研究者认为PDE7活性对T细胞并不重要。但多项研究表明,选择性PDE4/7抑制剂可抑制T细胞活性,抑制免疫反应[56]。BC54是一种PDE4/7选择性抑制剂,能促进慢性淋巴细胞白血病细胞凋亡,且效果要好于咯利普兰(PDE4抑制剂)和PDE7抑制剂BRL 50481。BRL 50481[57]也可诱导细胞凋亡,与其他PDE抑制剂联合使用抗肿瘤效果增强,如BRL 50481单独作用于CD8+T淋巴细胞、单核以及肺巨噬细胞时,CD8+T淋巴细胞增殖无变化,且肿瘤坏死因子α(tumor necrosis factor-α,TNF-α)生成仅降低2%~11%,但与咯利普兰联用后,明显增强咯利普兰对肿瘤细胞的增殖抑制作用,TNF-α生成也显著下降。另外,基于计算机模拟发现的小分子化合物S14和VP1.15也能通过抑制PDE7发挥抗炎作用[58]。
2.8 磷酸二酯酶10抑制剂
PDE10对cAMP和cGMP均有水解作用,但对cGMP水解作用更强[59]。PDE10A与肺癌、乳腺癌和结肠癌发生相关。抑制PDE10A可以抑制结直肠癌细胞生长,主要是由于cGMP/PKG信号通路激活阻断了β连环蛋白通路促进细胞核易位[60]。PDE10A在人非小细胞肺癌细胞中过表达,PDE10抑制剂PQ10和ADT-020能够迅速增加cGMP水平,激活PKG,抑制NSCLC细胞生长。PKG激活抑制β连环蛋白和丝裂原活化蛋白激酶(micogenactivated protein kinase,MAPK)信号传导诱导细胞凋亡是PDE10发挥抗肿瘤作用的重要机制[61]。目前PDE10抑制剂的研究主要集中在周围和中枢神经系统疾病,肿瘤相对较少。
2.9 其他PDE抑制剂
近年来,PDE在肿瘤中的作用研究逐渐增多,PDE及其抑制剂影响肿瘤细胞内cGMP和cAMP表达对肿瘤细胞的转移和增殖已经有很多报道,但关于PDE8,PDE9和PDE11在肿瘤中的作用研究仍较少。有研究者发现,PDE8在非分泌性肾上腺皮质癌中存在遗传缺陷,可能与肾上腺皮脂癌的易感性相关,但作用机制有待研究[62]。PDE9抑制剂WYQ-C36D可通过cGMP-CREB信号通路改善皮质酮诱导的神经毒性和抑郁行为[63]。另外,PDE9抑制剂在治疗肝纤维化中也具有较好疗效[64]。PDE9目前在肿瘤中的作用尚未见报道。PDE11与睾丸和肾上腺肿瘤的发生存在关联,但目前尚无PDE11的特异性抑制剂[65],其在肿瘤中的作用有待进一步研究。
3 磷酸二脂酶促肿瘤作用机制
从目前研究看来,PDE抑制剂发挥抗肿瘤作用主要是通过调控cAMP/cGMP水平影响下游信号分子实现。如cAMP可以抑制肾素-血管紧张素系统活性,降低肿瘤细胞中偏高的MAPK活性,而MAPK信号通路又可分为细胞外调节蛋白激酶(extracellular regulated protein kinases,ERK),应激活化蛋白激酶(c-Jun N-terminal kinase,JNK)和P38/MAPK,其中JNK、P38与细胞生长、凋亡和炎症有关;另一方面,cAMP/cGMP水平改变会使Bcl-2家族促凋亡蛋白易位到线粒体外膜,下调凋亡因子表达[66],如cAMP可降低细胞内Bcl-2和鼠双微粒体小体2(murine double minute-2,MDM2)水平,诱导肿瘤细胞凋亡。
CREB磷酸化,导致骨架重构,通过抑制PDE表达影响细胞迁移[67]。CREB是环核苷酸信号通路下游靶点,研究认为它与肿瘤凋亡、血管新生及转移有关[54,68]。Ca2+/CaM/CaM 激酶Ⅱ通路也可诱导CREB磷酸化[69],PDE1家族主要由Ca2+和CaM组成,因此抑制PDE1表达可抑制肿瘤进展。转录激活因子4(recombinant activating transcription factor 4,ATF4)是ATF/CREB家族的一员,ATF4在乳腺癌中过表达促使巨噬细胞向肿瘤细胞募集,促进肿瘤血管生成和细胞生长[70]。此外,血管内皮生长因子C可诱导肿瘤血管生成,促进肿瘤转移,而这些反应都需要CREB介导相关信号通路激活[71]。因此,cAMP/PKA/CREB通路是影响肿瘤细胞增殖和转移的重要信号通路。
PKG作为cGMP的主要靶分子,包括PKG1和PKG2亚型。PKG1参与结肠癌、乳腺癌和卵巢癌等多种肿瘤的发生发展,且PKG1α和PKG1β在不同肿瘤组织中呈现抗肿瘤和促肿瘤的不同效应[72-73]。β连环蛋白与肿瘤细胞失巢敏感性有关。研究发现,过表达PKG1可下调裸鼠结肠癌细胞移植瘤中β连环蛋白表达,抑制肿瘤血管生成[74]。TMPRSS2-EST转录因子家族相关基因(ETS-relatedgene,ERG)融合基因的异常激活是导致前列腺癌发展的主要事件,有研究团队发现,可溶性鸟苷酸环化酶(soluble guanylate cyclase,sGC)α 1和β1亚基在体内受到ERG的直接和特异性调节,而sGC是NO-cGMP信号传导的主要介质,与NO结合后,催化cGMP合成激活PKG促进肿瘤细胞增殖,靶向NO-cGMP信号通路是治疗前列腺癌的新策略[75]。此外,NO/sGC/cGMP/PKG信号通路能够下调p53介导的自发性凋亡,促进卵巢癌细胞DNA合成和增殖,具有促肿瘤效应[76]。PKG2还可通过抑制表皮生长因子受体信号通路转导,抑制卵巢癌发生发展[77]。
4 结语
PDE是一个庞大的家族,各亚型对环核苷酸的水解各有特点,基因和蛋白质组学研究结果和临床药物应用结果表明PDE可作为疾病治疗靶点的有效性和安全性。截至2018年,全球已经有40种PDE抑制剂作为治疗药物使用[12],近几年研究发现PDE在多种肿瘤中异常表达,对不同PDE家族各基因亚型调控肿瘤发生发展的机制进行研究,可为肿瘤治疗提供新靶点,将成为未来PDE研究的一大重要方向。但是,目前现有的具有抗肿瘤作用的PDE抑制剂大都还处于临床前研究阶段,部分临床研究才刚刚起步尚未取得明确结果,因此,新型高效和高选择性的PDE抑制剂的研发工作任重而道远。
