中华大仰蝽转录组学研究及SSR新标记开发
2022-02-15张丹丽李荣荣宋鲜梅卜文俊
李 敏, 张丹丽, 李荣荣, 雷 廷, 宋鲜梅, 卜文俊
(1.太原师范学院生物系, 山西晋中 030619; 2.南开大学生命科学学院昆虫学研究所, 天津 300071)
随着转录组测序技术的发展,对蝽类昆虫转录组测序越来越多,截止2021年1月,美国国家生物技术信息中心(NCBI)创建和维护的TSA(Transcriptome Shotgun Assembly)数据库收录了105种蝽类昆虫的转录组数据,主要来自于陆生吸血和植食性种类,如长红猎蝽Rhodniusprolixus、骚扰锥猎蝽Triatomainfestans、吸血锥猎蝽Triatomadimidiata、温带臭虫Cimexlectularius、茶翅蝽Halyomorphahalys、豆荚草盲蝽Lygushesperus、美国牧草盲蝽Lyguslineolaris、稻绿蝽Nezaraviridula、南瓜缘蝽Anasatristis等。对于其基因功能的挖掘,也主要集中于这些种类。Nevoa等(2018)利用高通量测序技术组装并描述了猎蝽科的锥蝽属Panstrongyluslignarius的唾液腺和脂肪体转录组产生的编码序列,发现在唾液腺的蛋白质家族中,脂钙素是最丰富的;脂肪体转录组显示了与代谢功能相关的蛋白质,提高了对血食性猎蝽的唾液腺和脂肪体功能的认识,并发现了在病媒和脊椎动物宿主之间相互作用中的重要分子。Denecke等(2020)以稻绿蝽N.viridula中肠为研究对象,利用转录组学和蛋白质组学的方法,建立了中肠4个部分的表达图谱,表明中肠前段(M1-M3区域)在消化和异种生物代谢中发挥作用,而中肠最后段(M4区域)富含跨膜蛋白,为后续研究稻绿蝽的中肠分子生理奠定了基础。对于水生、捕食性蝽类,目前只检索到TSA数据库中有绒盾大仰蝽N.glauca转录组测序数据,但是并未有详细的文献报道。
同源异型基因(homeotic genes)是调控生物体型生长发育的一类基因家族,该基因家族因含有一段长约180 bp高度保守的DNA序列,即同源异型盒(homeobox),也称为Hox基因。Hox基因在真核生物中广泛存在,通常成簇分布,在胚胎发育中对于体节和组织特征的决定以及器官的形成具有重要的调控作用(Erikssonetal., 2010; Feiner and Wood, 2019)。Hox基因最早在果蝇中发现,分为ANTP-C(antennapediacomplex)基因簇和BX-C(bithoraxcomplex)基因簇两大类,ANTP-C基因簇包括labial(lab),proboscipedia(pb),Deformed(Dfd),Sexcombsreduced(Scr),Antennapedia(Antp);BX-C基因簇包括Ultrabithorax(Ubx),abdominal-A(abd-A)和abdominal-B(abd-B)等基因(McGinnis and Krumlauf, 1992; Gehringetal., 2009)。
本研究拟采用高通量测序技术对中华大仰蝽进行转录组测序,建立中华大仰蝽转录组数据,并通过本地ncbi-blast-2.1软件对其Hox基因进行鉴定,为进一步研究其捕食行为及仰泳仿生学机制提供分子生物学依据;利用软件MISA(Beieretal., 2017)基于中华大仰蝽转录组unigenes进行SSR新标记挖掘和筛选,通过毛细管电泳进行多态性检测,为后期研究中华大仰蝽遗传多态性及基因图谱构建提供一种方便而快捷的途径,同时推动中华大仰蝽种质资源的保护。
1 材料与方法
1.1 供试昆虫
本研究试虫中华大仰蝽采自山西省太原市尖草坪区上兰村水文站(38°0′11″N, 112°26′28″E),去掉腹部后置于RNAlater(索莱宝Solarbio)样本保存液中,带回实验室放-80℃低温冰箱保存。中华大仰蝽地理种群信息见表1。
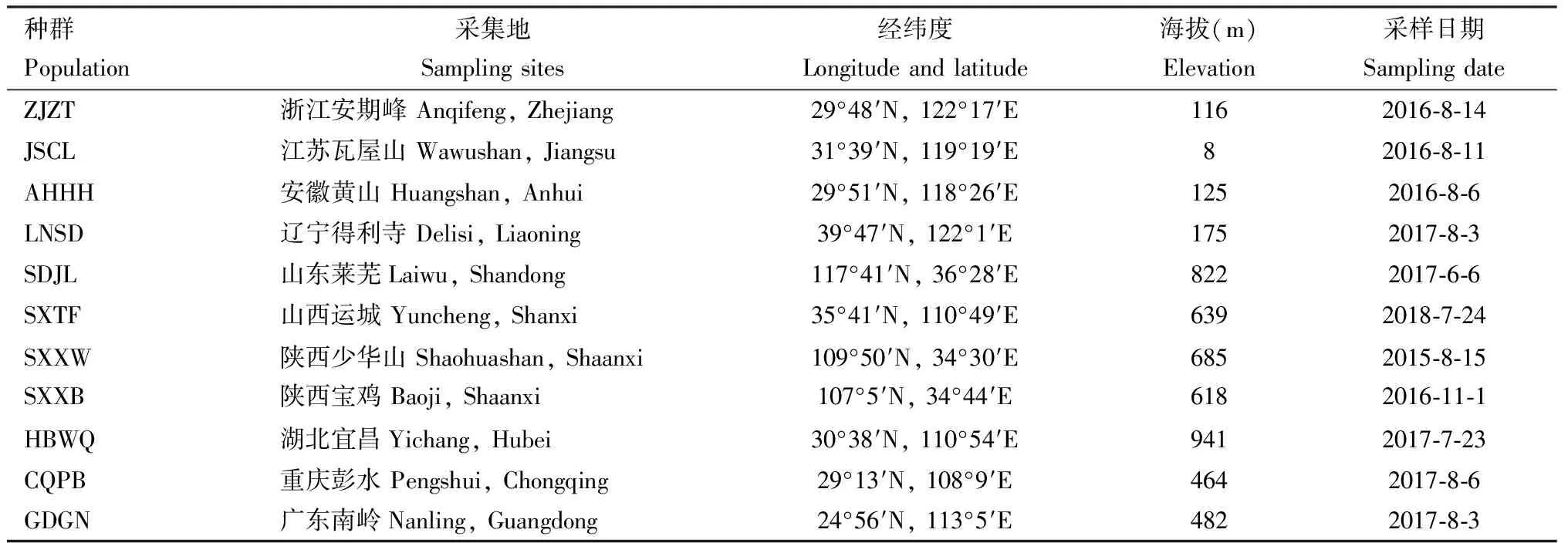
表1 中华大仰蝽地理种群样本信息
1.2 总RNA提取、cDNA文库构建及测序
用Trizol法提取中华大仰蝽的总RNA,总RNA质量以紫外分光光度计和琼脂糖凝胶电泳检测。样品检测合格后合成cDNA第1链,随后加入dNTPs、DNA Polymerase I、RNaseH和缓冲液合成cDNA第2链,并纯化双链cDNA。纯化的双链cDNA,先进行末端修复,加A尾并连接相应的测序接头,再用AMPureXP beads选择其片段大小,最后通过桥式PCR扩增,富集得到cDNA文库。文库构建完成后,使用Agilent 2100对文库质量进行评估,文库检测合格后采用第2代测序技术,基于Illumina NextSeq500测序平台,对文库进行双末端测序。
1.3 测序数据拼接和组装
对中华大仰蝽转录组测序得到原始序列(raw reads),通过接头污染去除和质量过滤获得clean reads。同时计算Q20(碱基识别准确率在99%以上的碱基所占百分比),Q30(碱基识别准确率在99.9%以上的碱基所占百分比)和GC含量。基于DBG(De Bruijn Graph)拼接原理,使用Trinity v2.0.2软件(k-mer 25 bp)对高质量序列(clean reads)进行拼接,获得转录本序列。
1.4 Unigenes的聚类与功能注释
将Trinity v2.0.2拼接得到的每一条transcript与参考nr数据库进行blastx比对(E-value<1e-5)。将比对至相同GI号的transcript归为同一unigene。对聚类得到的unigenes比对nr, NCBI, Swiss-Prot, GO, eggNOG和KEGG数据库进行基因功能注释。
1.5 Hox基因的筛选和鉴定
通过NCBI(http:∥www.ncbi.nlm.nih.gov/)的Protein数据库下载黑腹果蝇Drosophilamelanogaster的Hox基因家族序列,利用本地软件ncbi-blast-2.11.0中makeblastdb程序建立黑腹果蝇Hox蛋白序列参考数据库。以中华大仰蝽转录组unigenes为查询序列,利用ncbi-blast-2.11.0中blastx程序与所建本地参考数据库进行比对(E-value<1e-5),然后使用perl脚本在结果中筛选相似度大于50%的序列,获得中华大仰蝽的Hox基因序列。
同样方法下载半翅目豌豆长管蚜Acyrthosiphonpisum、鞘翅目赤拟谷盗Triboliumcastaneum和鳞翅目家蚕Bombyxmori的Hox蛋白序列,利用MEGA X(Kumaretal., 2018)软件基于p-distance模型和最小进化法(minimum evolution method, ME)构建Hox蛋白序列的系统树。
1.6 SSR引物设计与引物初筛
用MISA软件对中华大仰蝽转录组unigenes进行SSR位点扫描。选择二核苷酸至六核苷酸不同重复序列的SSR位点,用SSRHunter v1.3软件(Li and Wan, 2005)截取SSR位点上下游150 bp,利用Primer Premier 5.0(Lalitha, 2000)设计引物。引物设计遵循标准:引物序列长度为18~25 bp,预计扩增产物长度在100~350 bp,GC含量为40%~60%之间,退火温度为55~65℃,正反向引物的退火温度值相差不大于3℃,引物末端禁用A和T(Blairetal., 2009)。共设计引物29对(表2),由上海派森诺生物科技股份有限公司合成。
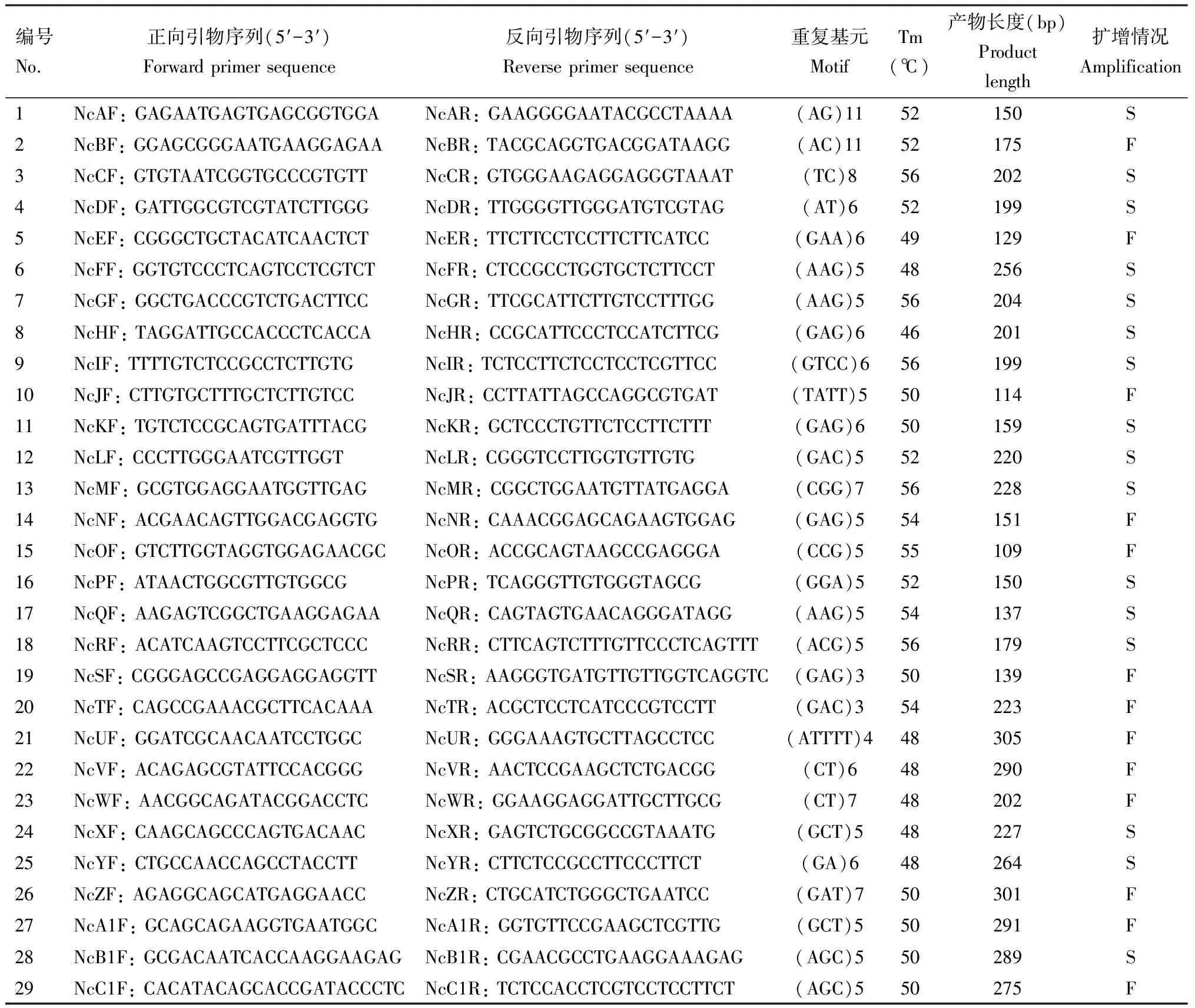
表2 基于中华大仰蝽转录组数据鉴定的SSR引物信息
利用通用型基因组提取试剂盒(康为世纪)按照说明书步骤提取中华大仰蝽浙江安期峰(ZJZT)、江苏瓦屋山(JSCL)和安徽黄山(AHHH)3个地理种群(表1)总基因组DNA,然后用NanoDrop 2000分光光度计(Thermo Scientific)检测基因组DNA浓度,将提取的基因组DNA在-20℃保存,用于SSR引物初筛。PCR反应体系(20 μL): 基因组DNA模板(130 ng/μL)1 μL, 2×Taq Master Mix 10 μL,正反向引物(10 μmol/L)各1 μL, 加ddH2O至20 μL。PCR扩增程序: 94℃预变性5 min; 94℃变性30 s, 最佳退火温度30 s, 72℃延伸30 s, 30次循环; 72℃再延伸5 min, 4℃保存。PCR产物用3%琼脂糖凝胶电泳,采用PBR 322 DNA/Msp Ⅰ Maker作为分子量标准,用凝胶成像系统(Bio-Rad)的Image Lab软件拍照,进行结果分析。
1.7 SSR引物复选及基因分型
从16对SSR引物中选择扩增条带单一且清晰的5对引物(NcAF/NcAR, NcCF/NcCR, NcKF/NcKR, NcLF/NcLR和NcQF/NcQR)(表2),在其正向引物的5′端加FAM荧光标记,用未加标记的反向引物与5′ FAM的正向引物对选自代表中国大陆的辽宁得利寺(LNSD)、山东莱芜(SDJL)、山西运城(SXTF)、陕西少华山(SXXW)、陕西宝鸡(SXXB)、湖北宜昌(HBWQ)、重庆彭水(CQPB)和广东南岭(GDGN)7个地理种群的样本(表1)分别进行PCR扩增,进行SSR引物复选、基因分型以及多态性验证。PCR反应体系(20 μL): 基因组DNA模板(15 ng/μL)1 μL, dNTPs(2.5 mmol/L)0.5 μL, Taq 酶0.5 μL, 正向荧光引物(10 μmol/L)1 μL, 反向普通引物(10 μmol/L)1 μL, 10×Buffer 2 μL, ddH2O 14 μL。PCR扩增程序: 95℃ 5 min; 95℃ 30 s, 62-52℃ 30 s, 10个循环; 72℃ 30 s, 95℃ 30 s, 52℃ 30 s, 25个循环; 72℃ 30 s, 72℃ 7 min, 4℃保存。扩增PCR产物用3730XL DNA Analyzer(ABI,美国)进行毛细管电泳检测。检测结果用GeneMarker v2.2.0软件读取,获得毛细管电泳的峰图和每个信号峰的片段长度。利用Cervus 3.0软件分析各个SSR位点的等位基因数(Na)、观测杂合度(Ho)、期望杂合度(He)和多态信息含量(PIC)。
2 结果
2.1 中华大仰蝽转录组测序、拼接和组装
运用Illumina NextSeq500测序平台对中华大仰蝽转录组测序,获得36 675 702条原始reads;碱基错误率为0.01%;Q20碱基比例为90.22%;Q30碱基比例为83.12%。数据过滤后,获得的clean reads平均数为34 782 282条(NCBI SRA数据库登录号: SRR13259254),组装到37 801条unigenes,总长度为25 517 069 bp,最大长度为23 542 bp,平均长度为675 bp。N50和N90分别为913 bp和304 bp,所对应序列数分别为7 888条和27 612条。GC百分比含量为52.92%。所有37 801条unigenes序列用于后续的SSR搜索。
2.2 中华大仰蝽转录组unigenes的功能注释
注释到nr数据库的unigenes最多,为36 474条,占96.49%;32 470条unigenes注释到Swiss-Prot数据库,占85.89%;27 781条unigenes注释到GO数据库,占73.49%;35 079条unigenes注释到eggNOG数据库,占92.80%;5 638条unigenes注释到KEGG数据库,占14.91%。在所有数据库中都能被注释到的unigenes为4 542条,占12.02%(表3)。
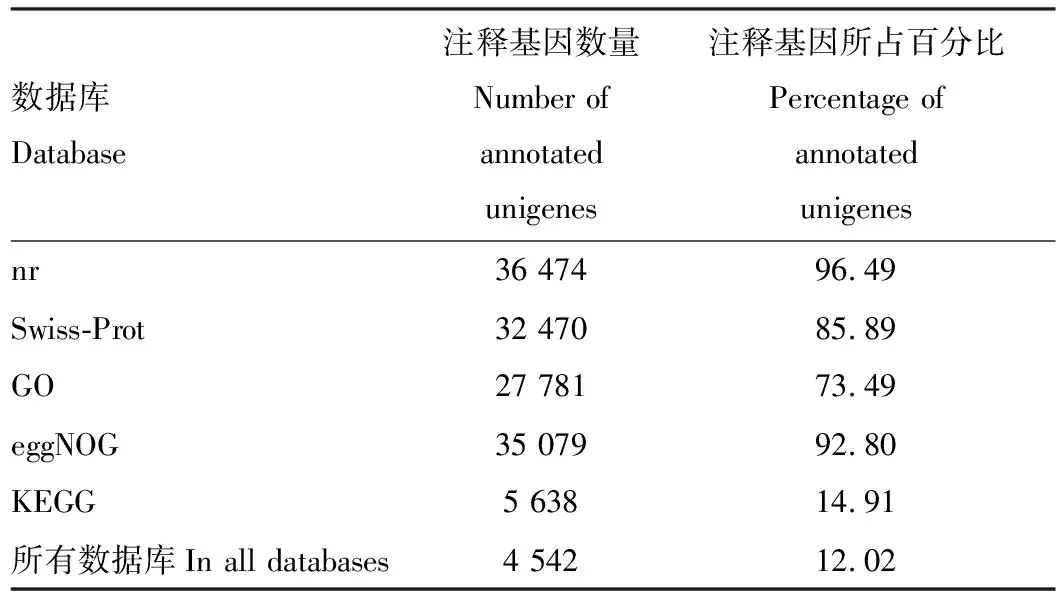
表3 中华大仰蝽转录组unigenes在5个数据库中的注释结果统计
GO注释结果表明,27 781条unigenes共得到279 368条功能注释,分为三大类,即生物学过程(biological process)、细胞组分(cellular component)和分子功能(molecular function)。其中生物学过程注释的序列数最多为119 086条,包括24个功能条目,其次是细胞组分注释的序列为118 137条,包括21个功能条目,注释序列最少的是分子功能为41 425条,包括17个功能条目(图1)。在生物学过程分类中,注释到细胞进程(cellular process)和单有机体进程(single-organism process)的序列数最多,分别为19 242和17 044条。在细胞组分中,注释到细胞(cell)和细胞部分(cell part)的序列占主导地位,分别为20 834和20 832条。在分子功能分类中,注释到结合(binding)和催化活性(catalytic activity)的序列数最多,分别为19 959和11 556条。注释的序列超过10 000条的功能条目共有11个,分别是细胞、细胞部分、结合、细胞进程、细胞器(organelle)、单有机体进程、代谢进程(metabolic process)、生物调节(biological regulation)、细胞器部分(organelle part)、催化活性和细胞膜(membrane)。
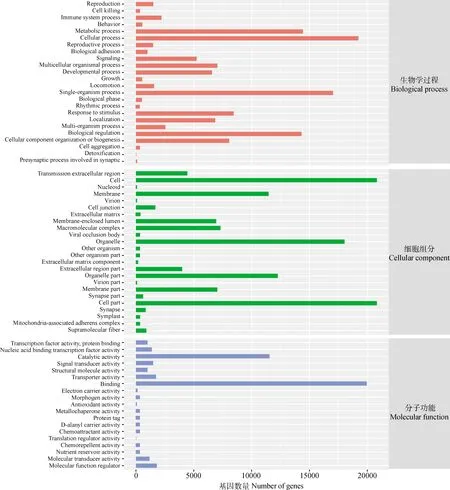
图1 中华大仰蝽转录组unigenes的GO功能注释
eggNOG的注释结果如图2所示。37 801条unigenes共得到41 259条注释,分为25类,数量最多的是注释到S类未知功能(function unknown),有6 205条;其余依次是T类信号转导机制(signal transduction mechanisms),有6 159条;R类仅一般功能预测(general function prediction only),有6 035条;K类转录(transcription),有3 840条;O类翻译后修饰、蛋白质转换、伴侣(posttranslational modification, protein turnover, chaperones)有3 147条;U类细胞内运输、分泌和囊泡运输(intracellular trafficking, secretion, and vesicular transport)有2 183条;其余的都低于2 000条;X类未定义的(undetermined)为0条。
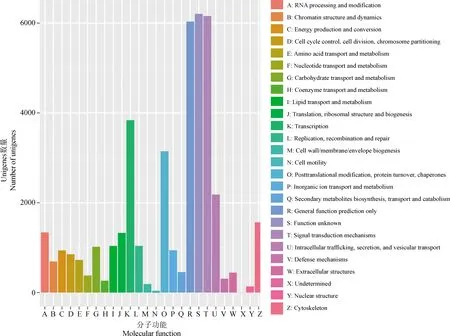
图2 中华大仰蝽转录组unigenes的eggNOG功能注释分类图
KEGG代谢通路富集分析结果表明,共有5 638条unigenes参与到新陈代谢(metabolism)、遗传信息处理(genetic information processing)、环境信息处理(environmental information processing)、细胞进程(cellular process)、人类疾病(human disease)和有机系统(organism system)这六大类生化代谢通路中(图3)。其中有机系统通路中基因最多(2 009条),它们主要参与内分泌系统(endocrine system)和免疫系统(immune system)等过程,分别为551条和410条。在这38组代谢通路子类别中,信号转导(signal transduction)通路的基因最多,为1 142条。这些代谢通路相关的基因分布于已知的245个代谢通路,其中富集最多的10条通路分别是核糖体(ribosome)、RNA运输(RNA transport)、内吞作用(endocytosis)、内质网中的蛋白质加工(protein processing in endoplasmic reticulum)、嘌呤代谢(purine metabolism)、剪接体(spliceosome)、氧化磷酸化(oxidative phosphorylation)、PI3K-Akt信号通路(PI3K-Akt signaling pathway)、泛素介导的蛋白水解作用(ubiquitin-mediated proteolysis)和嘧啶代谢(pyrimidine metabolism)。
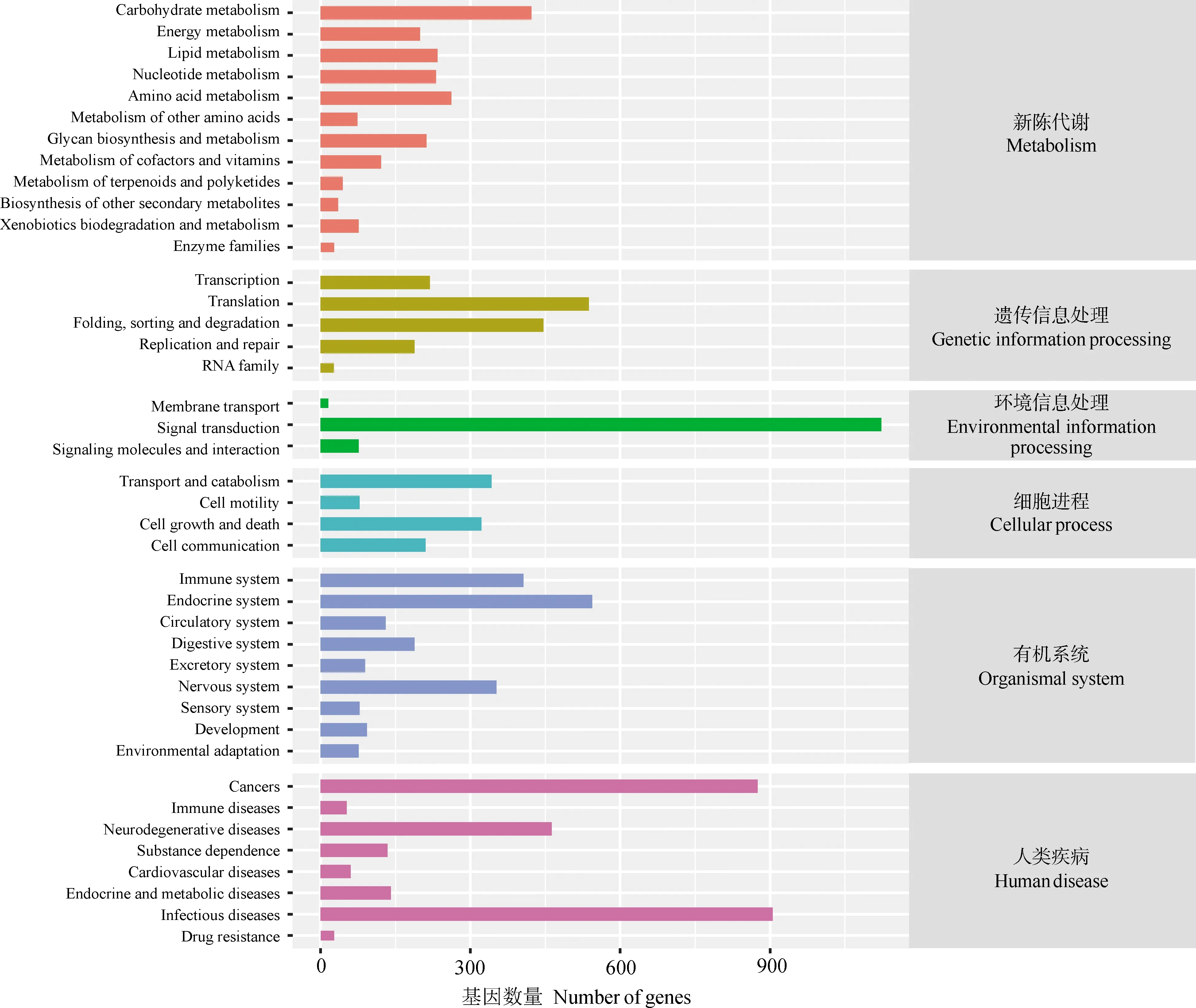
图3 中华大仰蝽转录组unigenes的KEGG通路分析
2.3 Hox基因家族的归类
中华大仰蝽转录组中共注释到17条Hox基因序列,包含了所有主要的8种Hox基因:lab1条(unigene: c23840_g1_i2),pb2条(unigenes: c57238_g1_i1, c14084_g1_i1),Dfd1条(unigene: c48312_g1_i1),Scr1条(unigene: c25114_g7_i1),Antp3条(unigenes: c25114_g1_i5, c25114_g6_i1, c25114_g3_i3),Ubx1条(unigene: c25114_g2_i1),abd-A1条(unigene: c25114_g5_i1)和abd-B7条(unigenes: c17276_g1_i1, c2333_g1_i1, c7432_g1_i2, c22062_g1_i1, c7763_g1_i1, c76858_g1_i1, c22143_g1_i1)(图4)。
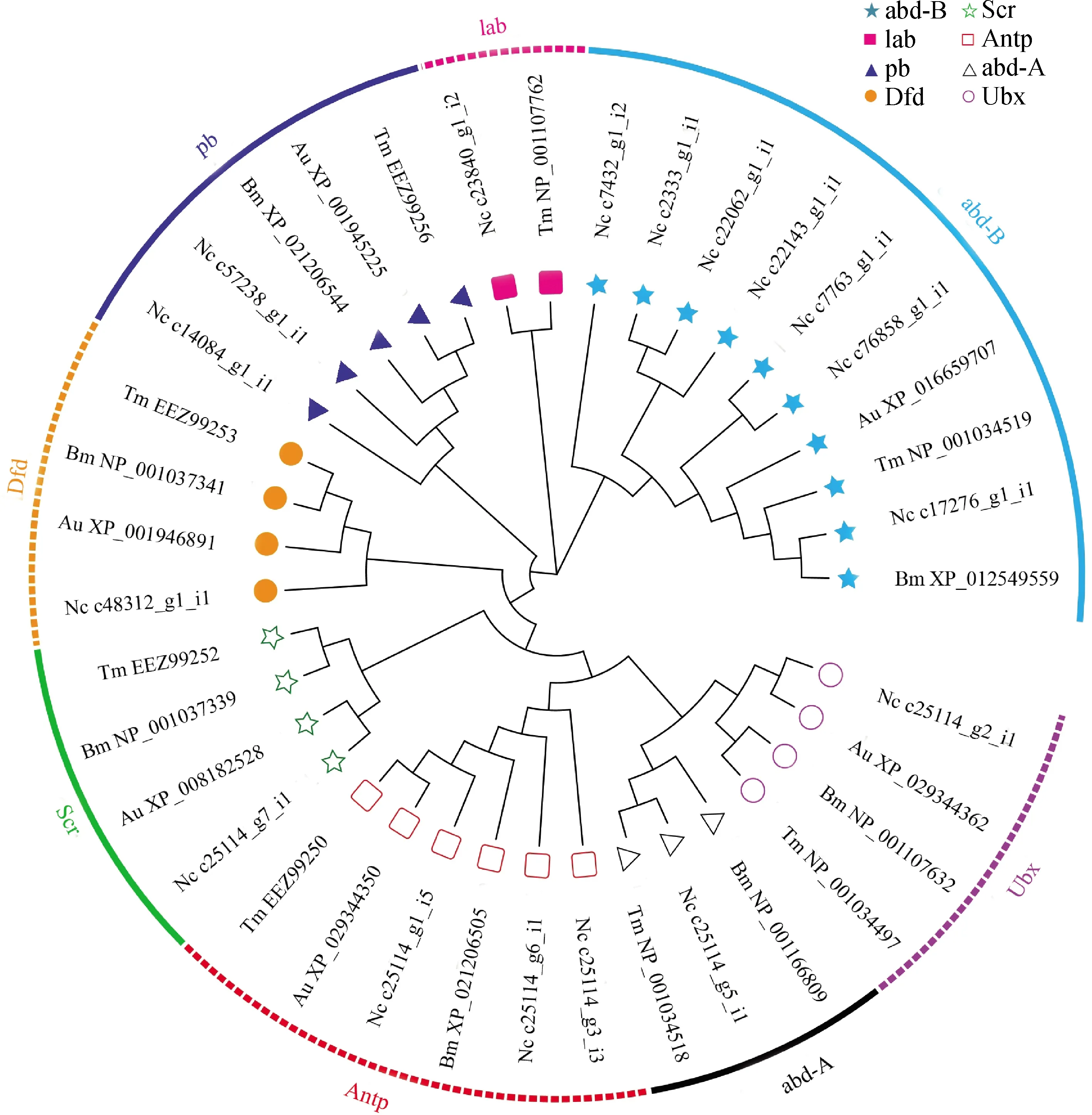
图4 最小进化法构建的基于氨基酸序列的Hox蛋白系统发生树
2.4 SSR分子标记开发与验证
中华大仰蝽转录组unigenes序列经MISA软件搜索,得到3 124个SSR位点,占总unigenes数量的8.26%,分布于2 671条unigenes序列中,发生频率(含有SSR位点的unigenes数量与总unigenes数量的比值)为7.07%。含1个以上SSR位点的unigenes序列350条,含不同重复类型SSR位点的unigenes序列171条。长度大于20 bp的SSR位点有245个,占总数的7.84%,长度在12~20 bp的SSR位点有1 859个,占总数的59.51%,其中20 bp以上的低级重复基元有237个,占20 bp以上SSR位点数的96.73%。从分布情况看,中华大仰蝽转录组unigenes中每8 168 bp就出现一个SSR位点,即平均距离。从中华大仰蝽转录组中共搜索到6种核苷酸重复类型,不同重复类型差异较大,重复基元主要以单、二和三核苷酸为主,有3 095个,占中华大仰蝽SSR位点总数的99.07%。出现数量最多的是单核苷酸重复类型,占总SSR位点的53.49%,其次是三核苷酸重复类型,占总SSR位点的33.00%,数量最少的是六核苷酸重复类型,只占总SSR位点的0.03%。不同核苷酸重复基元的重复次数有所差异,单核苷酸重复基元的重复次数主要集中在10~13次,二核苷酸重复基元的重复次数主要集中在6~8次,三核苷酸重复基元的重复次数主要集中在5~7次,四、五、六核苷酸重复基元的重复次数主要以5次为主(表4)。共有33种重复基元出现,单、二、三、四、五以及六核苷酸重复基元的种类分别是2, 4, 10, 14, 2和1种。其中出现频率最多的重复基元是A/T,其次是AGG/CCT。单核苷酸重复基元中,A/T为优势基元,占单核苷酸重复的88.51%;二核苷酸重复基元中,出现频率最多的是AC/GT,占二核苷酸重复的37.15%,其次是AG/CT,占二核苷酸重复的36.39%;三核苷酸重复基元中,出现频率最高的为AGG/CCT,占三核苷酸重复的24.25%;四、五和六核苷酸重复基元类型数量最少,总计占总SSR数量的0.91%(表5)。
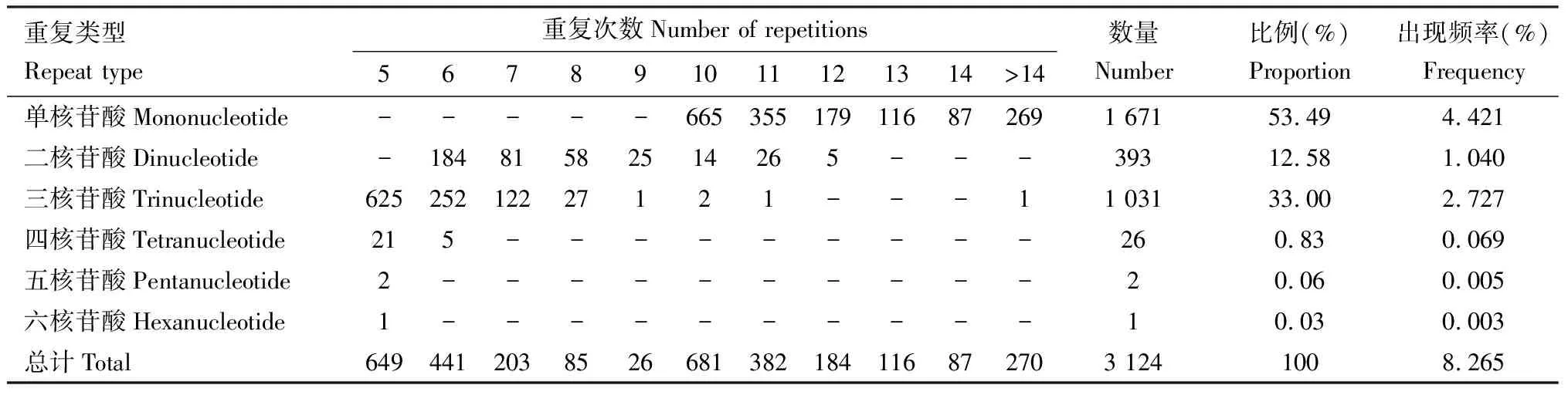
表4 中华大仰蝽转录组SSR位点的数量与分布
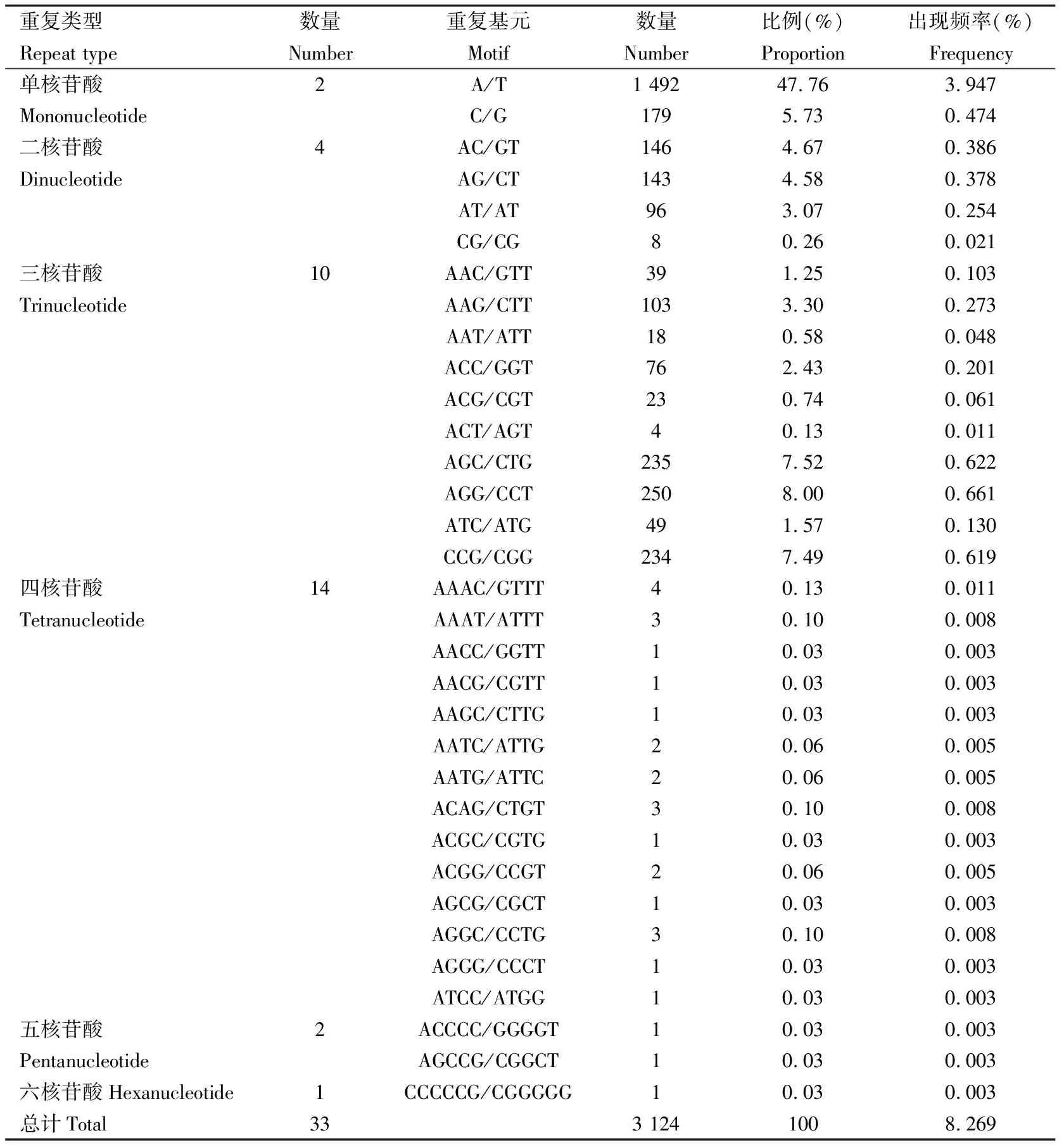
表5 中华大仰蝽转录组SSR重复基元的分布特征
PCR扩增结果表明,29对SSR引物中有16对可以成功扩增出目的条带,扩增效率为55.17%,有13对引物的扩增结果与预期产物片段不符。随机选取2对引物对3个地理种群进行PCR扩增,结果显示引物NcB1F/NcB1R和NcAF/NcAR均可以扩增出目的条带(图5)。毛细管电泳及GeneMarker v2.2.0软件分析显示,在7个群体中扩增5个位点(NcAF/NcAR, NcCF/NcCR, NcKF/NcKR, NcLF/NcLR和NcQF/NcQR),除NcQF/NcQR外,均有明确且符合预期的等位基因扩增产物。位点NcAF/NcAR等位基因数(Na)为2,多态信息含量(PIC)为0.375,在群体分析中所能提供的遗传信息较少;NcCF/NcCR, NcKF/NcKR和NcLF/NcLR等位基因数分别为13, 18和14,多态信息含量(PIC)分别为0.870, 0.902和0.857,均大于0.5(表6),说明中华大仰蝽微卫星的等位基因分布较丰富,可以用于种群差异性分析(Botsteinetal., 1980)。

图5 中华大仰蝽3个地理种群SSR引物NcB1F/NcB1R(A)和NcAF/NcAR(B)的PCR检测结果
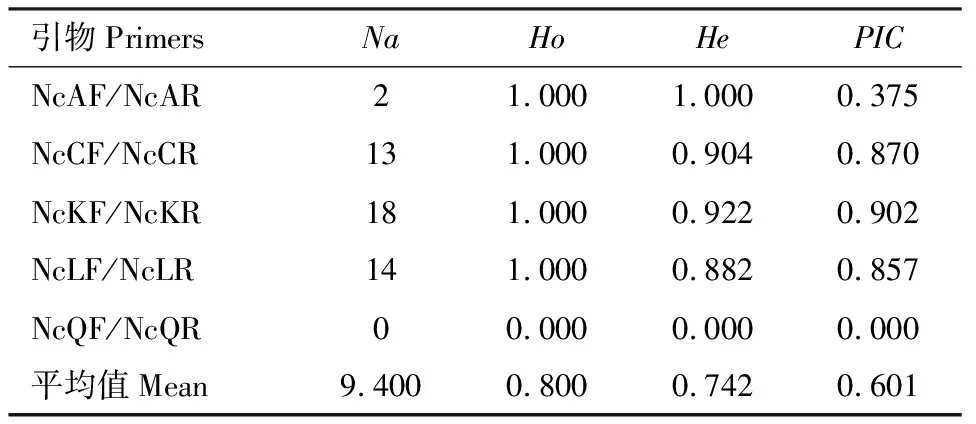
表6 中华大仰蝽7个地理种群转录组5个微卫星位点的遗传多样性
3 讨论
3.1 中华大仰蝽转录组
由于目前中华大仰蝽全基因组尚未公布,其各相关基因信息无法得知,导致中华大仰蝽基因功能研究进展缓慢。本研究通过RNA-seq技术对中华大仰蝽转录组进行测序,并进行denovo组装与分析,共获得34 782 282条clean reads,样本Q30碱基比例不低于83.12%,unigenes 37 801条,N50为913 bp。据报道,样本数据的Q30碱基比例不小于80%,测序质量较好;N50值越大说明序列拼接的完整性越好。可见本试验获得的测序数据质量较好,保证了转录组分析的准确性及重要功能基因挖掘的可能性。
根据与nr和Swiss-Prot蛋白质数据库的比对结果,36 474条unigenes(96.49%)比对至nr数据库,85.89%的unigenes被比对至Swiss-Prot数据库(表3),说明本次Illumina测序获得了大量在中华大仰蝽中表达的不同基因。GO数据库可以全面描述中华大仰蝽基因和基因产物的属性。本研究在GO数据库中共得到279 368条功能注释,分为三大类,其中注释到生物学过程的unigenes数目最多、其次为注释到细胞组分的,注释到分子功能的unigenes最少(图1),这与已报道的蠋蝽Armachinensis(Zouetal., 2013)、大垫尖翅蝗Epacromiuscoerulipes(金永玲等, 2015)、绿豆象Callosobruchuschinensis(郑海霞等, 2018)、中国真龙虱Cybisterchinensis(Hwangetal., 2018)以及麦红吸浆虫Sitodiplosismosellana(蒋月丽等, 2020)等相同。而黄曲条跳甲的转录组GO分类结果则是注释到分子功能的unigenes数目最多,其次为注释到生物学过程的,注释到细胞组分的unigenes最少(贺华良等, 2012),这可能与不同的昆虫有关。利用eggNOG数据库对中华大仰蝽unigenes进行基因功能分类(图2),可从组学水平上找寻直系同源体,预测未知ORF的生物学功能。研究表明基于Nanopore长读段测序数据在全长ORF预测和鉴定新基因方面具有显著优势(杜宇等, 2020),为下一步完善中华大仰蝽的组学信息、提高基因功能注释的准确性,提供了可靠思路。根据KEGG数据库对上述unigenes进行代谢通路分析,发现涉及245个具体的代谢通路分支,参与到中华大仰蝽的核糖体、RNA运输、内吞、内质网中的蛋白质加工、嘌呤代谢、剪接体、氧化磷酸化、PI3K-Akt信号通路、泛素介导的蛋白水解作用和嘧啶代谢等过程中(图3)。同时鉴定出了所有主要的8种Hox基因(lab,pb,Dfd,Scr,Antp,Ubx,abd-A和abd-B),为进一步大量挖掘中华大仰蝽生长发育过程中的重要表达基因,开展中华大仰蝽的基因克隆及功能验证等研究提供了基础数据。
3.2 中华大仰蝽转录组SSR引物设计
设计并进行PCR扩增的29对引物中,16对可以扩增出目的条带,但部分目的条带不是很清楚,且出现非特异性条带。虽然目前无法确定它们多态性的高低,但是说明用于多态性检测和分析的SSR位点的数量相当可观。SSR位点扩增失败可能是由于本研究所设计引物的SSR序列在基因组中的覆盖率很低,造成扩增产物很少以至于无法被检测到,而对于一些引物的扩增产物具有大量非特异性条带的情况,很可能是因为这些SSR位于同源基因序列上的缘故(魏丹丹等, 2014)。
3.3 中华大仰蝽SSR的特性
通过生物信息学方法,从中华大仰蝽转录组数据库37 801条unigenes中发掘出3 124个SSR位点,位点出现频率为8.26%,比黄粉虫Tenebriomolitor(1.67%)(Zhuetal., 2013)、云南切梢小蠹Tomicusyunnanensis(1.29%)(袁远等, 2014)、桔小实蝇Bactroceradorsalis(4.23%)(魏丹丹等, 2014)和扶桑绵粉蚧Phenacoccussolenopsis(5.79%)(罗梅等, 2014)出现频率要高,比灰飞虱Laodelphaxstriatellus(16.67%)(Zhangetal., 2010)和黑翅土白蚁Odontotermesformosanus(9.98%)(Huangetal., 2012)出现频率要低。分析原因,除了由于SSR搜索方法或标准有所差异外,最根本的原因可能是物种本身的差异。
中华大仰蝽转录组SSR的种类较多,一至六核苷酸重复都有出现,重复类型主要为单核苷酸重复,其次是三核苷酸重复(表4),这与扶桑绵粉蚧、黄粉虫和褐飞虱Nilaparvatalugens(刘玉娣和侯茂林, 2010)SSR重复类型是一样的。而在粘虫Mythimnaseparata(胡艳华等, 2015)及近缘的蠋蝽(Zouetal., 2013)转录组中SSR重复类型主要为单核苷酸重复,其次是二核苷酸重复;黑翅土白蚁O.formosanus转录组中SSR重复类型主要为二核苷酸重复,其次是三核苷酸重复;齿缘刺猎蝽Sclominaerinacea(黎东海和赵萍, 2019)转录组中SSR重复类型主要为三核苷酸重复,其次是二核苷酸重复。由此可见不同物种间SSR的重复类型存在差异。
中华大仰蝽SSR重复基元主要以单、二和三核苷酸为主,有3 095个,占中华大仰蝽SSR位点总数的99.07%。Dreisigacker等(2004)认为低级基元,包括单、二、三核苷酸重复基元普遍比高级基元的SSR多态性高。Temnykh等(2001)的研究表明,SSR的长度是影响多态性的重要因素。当SSR长度在20 bp及20 bp以上时多态性较高,在12~20 bp之间的SSR多态性中等,长度在12 bp以下时多态性极低。本研究发现长度大于20 bp的SSR位点有245个,占SSR位点总数的7.84%,长度在12~20 bp的SSR位点有1 859个,占SSR位点总数的59.51%。此外,本研究中20 bp以上的低级重复基元较多,共237个,占20 bp以上SSR位点数的96.73%。
等位基因数和杂合度与SSR位点的多态性成正相关。多态信息含量(PIC)是衡量微卫星位点基因多态性高低的重要指标。PIC>0.5具高度多态性,0.25
