复方玉红栓的质量标准研究
2022-02-12胡叶帅唐晓萌王志君黄月英王晓君宋洪杰海军军医大学长海医院药学部上海200433
胡叶帅,唐晓萌,王志君,黄月英,王晓君,宋洪杰(海军军医大学长海医院药学部,上海 200433)
复方玉红栓是长海医院特色医院制剂,处方由磺胺嘧啶、盐酸达克罗宁、苦参、槟榔、松香、紫草、白芷等中西药组成,临床使用有30 多年历史。该药具有消炎[1]、止痛[2]及止血[3]等作用,用于治疗混合痔内出血、内痔水肿脱垂、单纯内痔出血等,临床疗效显著,但目前质量标准仅对2 种化学药盐酸达克罗宁和磺胺嘧啶进行鉴别和含量测定,其他成分未制定质量标准,有待提升。本研究建立了苦参、松香、白芷的TLC 鉴别方法,同时建立测定磺胺嘧啶和盐酸达克罗宁含量的HPLC 方法,实现更好地控制本品质量、提高疗效稳定性和可靠性、增强临床使用的安全性目的。
1 材 料
1.1 仪器
岛津高效液相色谱仪(包括LC-20AD 泵、SPD-M20A 二极管阵列检测器、CBM-20A 控制器和LC solution 工作站,日本岛津公司);Goodsee-20E 型薄层成像系统(上海科哲生化科技有限公司);BT124S 电子天平(北京赛多利斯仪器有限公司);UV2550 紫外分光光度计(日本岛津公司);DL-720A 超声波清洗器(上海之信仪器有限公司);HWS24 型电热恒温水浴锅(上海一恒科技有限公司);硅胶G 薄层板(上海东方药品科技实业有限公司,规格10 cm×10 cm,批号:20190426);硅胶GF254薄层板(上海东方药品科技实业有限公司,规格10 cm×10 cm,批号:20141129)。
1.2 试药
复方玉红栓(本院自制,规格:每粒重1.4g,磺胺嘧啶25 mg、含盐酸达克罗宁5 mg;包装:7 粒/盒,批号:200 518、190 531、190 225),松香酸(批号A25F6C1,含量大于90%,上海源叶生物科技有限公司),磺胺嘧啶(批号100 026-201 404,含量99.7%),盐酸达克罗宁(批号100 423-201 102、含量99.8%),苦参碱(批号110 805-201 709,含量99.6%),白芷药材(批号120 984-200 602),苦参药材(批号121 019-201 604)均购自中国食品药品检定研究院;甲醇、乙腈为色谱纯,其余所用化学试剂均为分析纯,水为蒸馏水。
2 方法和结果
2.1 TLC 鉴别
2.1.1 白芷
取本品7 粒(批号200 518),加乙醇200 ml,水浴加热使溶解,放冷至室温,抽滤,滤液挥至无醇味后,加0.1 mol/L 盐酸溶液100 ml,加热使溶解,静置分层,取上层溶液,加0.1 mol/L 氢氧化钠溶液100 ml,萃取,取下层溶液,用盐酸调节pH 值至5,加石油醚(30~60 ℃)50 ml 萃取2 次[4],合并石油醚层挥干,残渣加乙醇2 ml 使溶解,作供试品溶液;同法制备缺白芷的阴性溶液;另取白芷药材0.5 g,同法制成对照药材溶液。取上述3 种溶液各10 μl,分别点于同一硅胶G 薄层板上,以石油醚(60~90 ℃)-乙醚(6∶5)为展开剂,展开,取出晾干,置紫外灯(365 nm)下检视。结果供试品色谱中,在与对照药材色谱相应的位置上,显相同颜色斑点,阴性对照在此相应位置无斑点。薄层色谱图见图1A。
2.1.2 松香
取本品1 粒(批号200518),加乙醇100 ml[5],水浴加热使溶解,冷藏1 h,取出过滤,取滤液挥干,残渣加乙醇2 ml 使溶解,取上清液作供试品溶液;同法制备缺松香的阴性溶液;另取松香酸对照品适量,加乙醇溶解制成每1 ml 含松香酸1 mg 的溶液,作对照品溶液。取上述3 种溶液各10 μl,分别点于同一硅胶GF254薄层板上,以石油醚(60~90 ℃)-乙酸乙酯-冰醋酸(8∶2∶0.1)为展开剂[6],展开,取出晾干,置紫外灯(254 nm)下检视。结果供试品色谱中,在与对照品色谱相应的位置上,显相同颜色斑点,阴性对照在此相应位置无斑点。薄层色谱图见图1B。

图1 复方玉红栓的TLC 图
2.1.3 苦参
取本品14 粒(批号200 518),加0.1mol/L 盐酸溶液100 ml,水浴加热使溶解,分取下层溶液,同法操作2 次合并下层溶液,加2 mol/L 氢氧化钠溶液调节pH 值至9,加三氯甲烷50 ml 萃取2 次[7],取三氯甲烷层挥干,残渣加乙醇2 ml 使其溶解,作供试品溶液;同法制备缺苦参的阴性溶液;另取苦参药材1 g,同法制成对照药材溶液;取苦参碱适量,加乙醇溶解制成每1 ml 含苦参碱0.5 mg 的溶液,作对照品溶液。取上述4 种溶液各10 μl,分别点于同一硅胶G 薄层板上,以环己烷-乙酸乙酯-二乙胺(5∶4∶0.5)为展开剂,展开,取出晾干,喷以稀碘化铋钾试液显色[8],置日光灯下检视。结果供试品色谱中,在与对照药材和对照品色谱相应位置上,显相同颜色斑点,阴性对照在此相应位置无斑点。薄层色谱图见图1C。
2.2 HPLC 法同时测定磺胺嘧啶和盐酸达克罗宁含量
2.2.1 色谱条件
SHIMADZU C18柱(150 mm×4.6 mm,5 μm);流动相为甲醇(A)-0.02 mol/L 磷酸二氢钾溶液(B),用磷酸调节pH 值至3.3,梯度洗脱(0~6.0 min,25 %B;6.0~25.0 min,50 % B;25.0~30.0 min,25 % B);流速:1.0 ml/min;柱温:室温;检测波长:280 nm;进样量:20 μl。
2.2.2 溶液制备
(1)对照品溶液:取磺胺嘧啶对照品适量,精密称定,加甲醇制成每1 ml 含磺胺嘧啶0.25 mg 的溶液,作为储备液1;取盐酸达克罗宁对照品适量,精密称定,加甲醇制成每1 ml 含盐酸达克罗宁0.25 mg的溶液,作为储备液2;精密量取储备液1 和储备液2 适量,加流动相稀释制成每1 ml 含磺胺嘧啶50 μg、盐酸达克罗宁10 μg 的混合对照品溶液。
(2)供试品溶液:取本品10 粒(批号200518),切碎,取约0.7 g(相当于磺胺嘧啶12.5 mg,盐酸达克罗宁2.5mg),精密称定,置50 ml 量瓶中,加甲醇25 ml,90 ℃水浴加热5 min,放冷,加甲醇稀释至刻度,摇匀,冷藏静置1 h,滤过,取续滤液放至室温,再精密量取续滤液5 ml,置25 ml 量瓶中,用流动相稀释至刻度、摇匀,作供试品溶液。
(3)阴性样品溶液:根据处方制备不含磺胺嘧啶和盐酸达克罗宁的阴性样品,再按(2)项下方法制成阴性对照溶液。
2.2.3 专属性试验
分别取“2.2.2”项下混合对照品溶液、供试品溶液和阴性对照液各20 μl 注入液相色谱仪,按“2.2.1”项下色谱条件进样测定,记录色谱图,结果见图2。结果表明,在混合对照品色谱相应位置上,供试品溶液色谱图中均具有相同保留时间的色谱峰,而阴性对照液在此处均无吸收峰,表明检验方法的专属性良好。
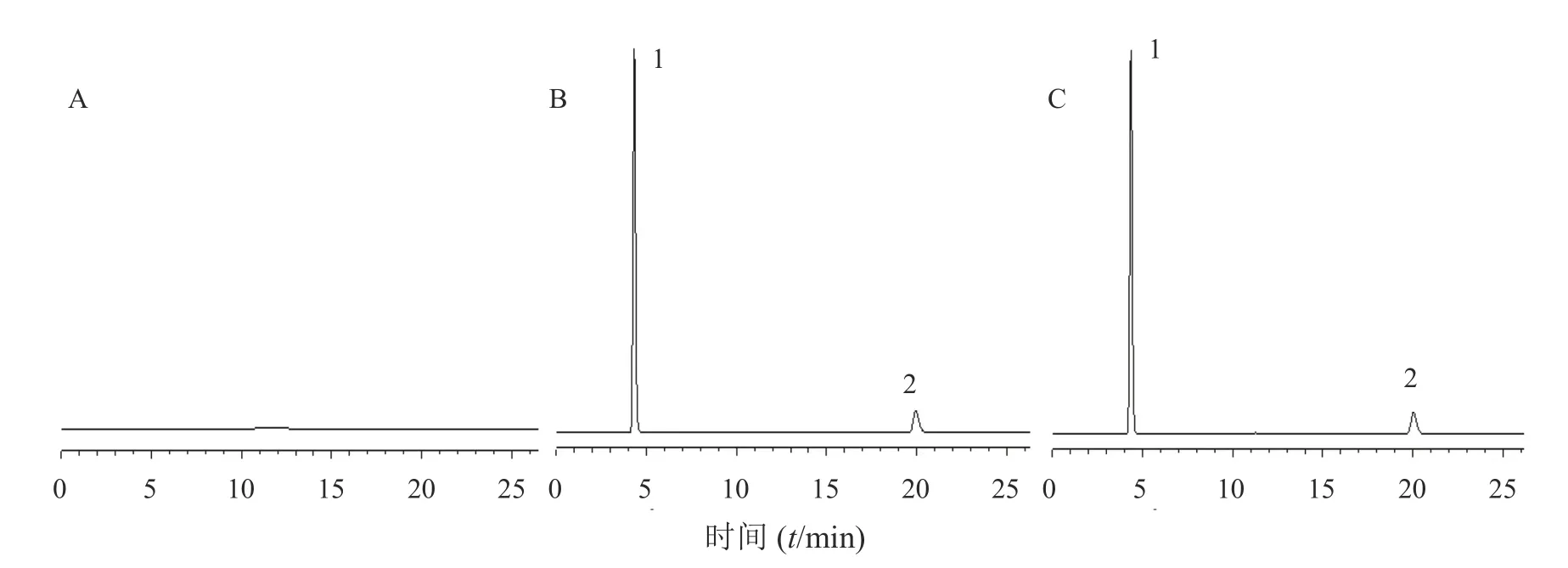
图2 复方玉红栓HPLC 色谱图
2.2.4 线性关系考察
分别精密量取“2.2.2”项下(1)的储备液1 和储备液2 适量,用流动相稀释,配制磺胺嘧啶系列浓度为12.40、24.80、49.60、74.40、99.20 μg/ml,盐酸达克罗系列浓度2.56、5.12、10.24、15.36、20.48 μg/ml的混合对照品溶液。按“2.2.1”项下色谱条件,进样20 μl,记录峰面积,以对照品浓度(C)为横坐标,峰面积(A)为纵坐标,进行线性回归分析,得到磺胺嘧啶和盐酸达克罗宁的回归方程分别为A盐酸达克罗宁=77 680c+44 018(r=0.999 9),线性范围2.56~20.48 μg/ml;A磺胺嘧啶=72 528C+2 862.9(r=0.999 9),线性范围12.40~99.20 μg/ml。结果表明磺胺嘧啶和盐酸达克罗宁在相应范围内线性关系良好。
2.2.5 精密度试验
精密吸取"2.2.2"项下(1)的混合对照品溶液20 μl,按"2.2.1"项下色谱条件重复进样6 次;同法操作,每天进样1 次,共进样6 d,分别记录,按峰面积计算得磺胺嘧啶和盐酸达克罗宁的日内精密度分别为0.07 %和0.58 %(n=6),日间精密度分别为1.60 %和1.65 %(n=6)。结果表明仪器精密度良好。
2.2.6 稳定性试验
分别取同一供试品溶液(批号200 518),在0、1、2、4、8、12 h 按"2.2.1"项下色谱条件进样,按峰面积计算得磺胺嘧啶和盐酸达克罗宁的RSD 分别为0.74 %和0.92 %(n=6),表明供试品溶液在12 h 内稳定。
2.2.7 重复性试验
取本品(批号200 518),按“2.2.2”项下(2)的方法制备供试品溶液6 份,按“2.2.1”项下色谱条件进样,记录峰面积,计算磺胺嘧啶和盐酸达克罗宁的RSD 分别为1.90 %和1.58 %(n=6),表明该方法重复性良好。
2.2.8 回收率试验
按处方工艺制备磺胺嘧啶和盐酸达克罗宁标示量为80%、100%、120%的3 种样品,按“2.2.2”项下(2)的方法制备供试品溶液各3 份,并按“2.2.1”项下条件测定,计算平均回收率,结果表明磺胺嘧啶和盐酸达克罗宁的平均回收率分别为(99.10±0.48)%、(99.54±0.68)%(n=9)。
2.2.9 含量测定
取3 个批号样品,分别按“2.2.2”项下(2)的处理方法制备供试品溶液,按“2.2.1”项下色谱条件测定峰面积,代入回归方程计算含量,结果见表1。
表1 磺胺嘧啶和盐酸达克罗宁的含量测定结果(±s,n=3)
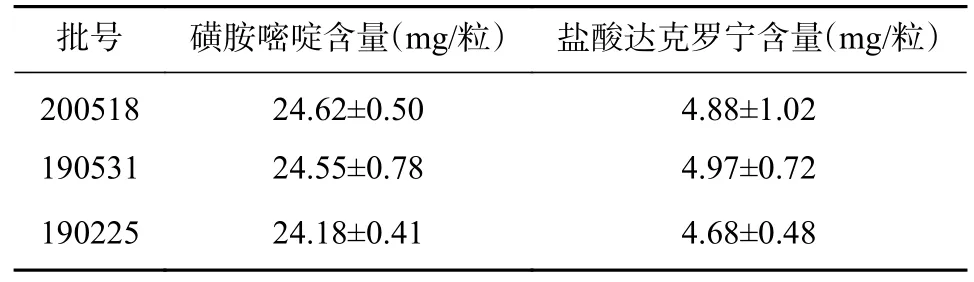
表1 磺胺嘧啶和盐酸达克罗宁的含量测定结果(±s,n=3)
3 讨论
3.1 薄层色谱鉴别
复方玉红栓是以混合脂肪酸甘油酯作为油脂性基质的栓剂,中药成分在处方中含量少,以原药材总量计仅为4 %,油脂性基质占比90 %,药物受基质影响很大,若操作过程中油脂性基质未完全除去,中药有效成分提取不完全,实验时易发生斑点拖尾甚至没有斑点显现,严重影响鉴别的专属性和灵敏度,所以在操作过程中去除干扰的油脂性基质和选择合适提取方法至关重要[9]。根据待测药材的成分特性,本研究在参考文献的基础上建立了白芷、松香、苦参3 种中药材的提取和TLC 鉴别方法,所建立的方法可以用于复方玉红栓制剂中3 种药材的鉴别。本研究还尝试建立紫草和槟榔的TLC 鉴别方法,但两者在处方中所占比例更少,大量混合脂肪酸甘油酯严重干扰两味药材的提取,因此尚未建立它们特征性的鉴别方法。
3.2 HPLC 法同时测定磺胺嘧啶和盐酸达克罗宁含量
目前尚未见有同时测定磺胺嘧啶和盐酸达克罗宁的研究报道。本研究采用甲醇-0.02 mol/L 磷酸二氢钾溶液(用磷酸调节pH 值至3.3)为流动相,建立了梯度洗脱条件,同时测定两者的含量。对色谱条件考察时发现,随着0.02 mol/L 磷酸二氢钾溶液比例增加,盐酸达克罗宁出峰时间提前,但峰形不对称影响测定准确性,经过摸索最终确定本实验洗脱比例[10]。研究中考察了两种成分的提取条件,发现不同的提取温度和时间影响提取效率,盐酸达克罗宁不稳定[11],遇热易发生降解,本实验摸索了水浴温度90 ℃,提取5 min 的条件,既保证了有效成分提取完全又防止了盐酸达克罗宁的降解。
本研究首次建立了松香、苦参、和白芷的薄层色谱鉴别方法,此方法操作性强,斑点显色清晰;用HPLC 法同时测定磺胺嘧啶和盐酸达克罗宁,此方法的准确性、重现性好,符合快速测定要求。本研究为该制剂全面控制药品质量和临床疗效提供了重要依据。
