半芳香尼龙PA12T的微波辅助聚合与表征
2022-02-12黄文蕊黄正强付鹏崔喆张晓朦庞新厂赵清香刘民英
黄文蕊,黄正强,付鹏,2,崔喆,2,张晓朦,庞新厂,2,赵清香,2,3,刘民英,2,3
(1 郑州大学材料科学与工程学院,河南 郑州 450001;2 河南省先进尼龙材料及应用重点实验室(郑州大学),河南 郑州 450001;3 中国石油和化工行业高性能尼龙工程塑料工程实验室,河南 郑州 450052)
半芳香尼龙因其优异的耐热性能,广泛应用于汽车、集成电路及电气等行业,需求量不断扩大,已经商业化的半芳香尼龙主要品种有PA 6T、PA 9T 和PA 10T等。为解决半芳香尼龙产物的熔融黏度高、从聚合装置中排出困难的工艺难题,目前普遍采用熔融制备低黏度预聚物,再进行固相后聚合的制备方法。该方法存在反应周期长、分子量分布宽、能耗高且易产生部分凝胶及色泽发黄等问题。
微波辅助反应是一种快速、绿色、高效的合成方法,在高分子合成领域日益受到人们的关注。在传统熔融聚合过程中,通常采用聚合釜壁加热方式,并通过机械搅拌促进传热。但该方法存在升温速度慢、受热不均匀的现象,导致聚合反应不均匀而产生分子量分布较宽的问题。而微波辅助聚合具有升温迅速、均匀等优点。研究表明,在能量输入相等的情况下,微波加热的升温速率比传统加热方式提高几个数量级,可以极为有效地缩短反应时间,提高制备效率,降低能量消耗。Imai 等报道了在反应体系中加入极性有机溶剂的微波辅助脂肪族尼龙聚合,特性黏数为0.5dL/g。Park等以多种芳香二胺和芳香二酸为单体,以−甲基吡咯烷酮(NMP)为溶剂,在家用微波炉中合成了一系列的全芳香尼龙,研究了催化剂的浓度对反应产物的影响。目前为止,关于半芳香聚酰胺的微波辅助聚合尚未见文献报道。
本文以尼龙12T(PA12T)预聚物为目标产物,以少量水为微波吸收剂,研究了微波辅助聚合条件对聚合产物特性黏数的影响,并对聚合产物的结构和热性能进行了表征。
1 实验
1.1 微波聚合原理
微波加热技术能通过吸波物质随微波电场快速运动,使体系快速均匀升温,其能耗仅为传统加热方式的几分之一或几十分之一。有文献报道,尼龙本身基本不吸收微波,不能通过微波直接作用使聚合反应体系升温,需要加入吸波介质吸收微波能,达到加热目的。本研究通过加入微波吸收剂−水来实现升温目的。图1是微波辅助聚合PA12T示意图。如图1 所示,聚合前PA12T 盐呈粉末状态,少量水分子均匀混合在PA12T 盐小颗粒之间,通过水分子吸收微波产生热能,水分子与PA12T 盐颗粒表面的碰撞运动使盐表面升温,通过热传导把热量传导到盐颗粒内部,使反应体系升温。在本反应体系中,水除了具有微波吸收剂的作用,还可以与PA12T 盐产生氢键作用,破坏PA12T 盐晶体,使PA12T盐在低于其熔点温度下发生聚合反应。
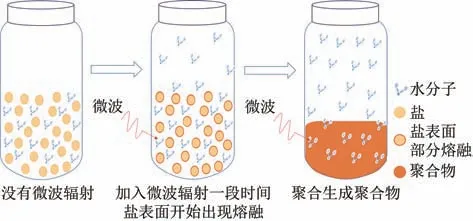
图1 微波辅助聚合尼龙示意图
1.2 实验原料
1,12−十二碳二元胺(C12DA),工业级,河南省君恒实业集团生物科技有限公司;对苯二甲酸(PTA),工业级,中国石化洛阳石化分公司;蒸馏水,自制;传统聚合PA12T样品,自制。
1.3 主要仪器
微波合成反应器,Monowave300,频率2.45GHz,最大功率850W,最高工作温度300℃,最高工作压力3MPa,奥地利安东帕(中国)有限公司;热重分析仪(TG),TGA55,美国TA 公司;差示扫描量热仪(DSC),TA Q2000,美国TA公司;红外光谱分析仪(FTIR),TENSORII,德国布鲁克公司;超导核磁共振仪(H NMR),Bruker DPX−400,德国布鲁克公司;乌氏黏度计,毛细管内径为0.9~1.0mm,黏度计常数为0.06619mm/s,上海宝山启航玻璃仪器厂。
1.4 PA12T盐制备
称取等摩尔比的C12DA 和PTA,量取反应物质量3倍的蒸馏水加入反应釜中,然后将C12DA和PTA加入反应釜中,用惰性气体置换釜内空气,密闭,升温至140℃,在搅拌下反应2h 后,停止加热,冷却至室温,抽滤,60℃真空下烘干12h,得到白色粉末状PA12T 盐。为273℃,90℃水中溶解度为3.5g。
1.5 微波辅助聚合
1.5.1 升温速率控制
称取4g干燥的PA12T盐放入30mL的微波反应瓶中,在反应瓶中加入不同量的去离子水,混合均匀,用高纯CO置换反应瓶中空气,把反应瓶放入微波反应器。设置升温程序,10min从室温快速升温到130℃,保温5min,10min 快速升温至170℃,再以10℃/min的升温速率升温至不同反应温度,并保温不同的时间后,降温至常温,取出产物,在80℃下烘干后得到块状PA12T。
1.5.2 微波功率控制
称取4g干燥的PA12T盐放入30mL微波反应瓶中,在反应瓶中加入0.17mL 去离子水,混合均匀后,以与1.5.1节相同程序升温至170℃,再以一定功率(20W、 30W、 40W、 50W、 60W、 80W、100W、150W、200W、250W)升温至210℃,保温60min,降温至常温,取出产物,在80℃下烘干后得到块状PA12T。
1.6 表征与测试
1.6.1 PA12T盐
FTIR表征:PA12T盐粉末与KBr研磨压片,测定傅里叶红外变换光谱(FTIR)。
热重(TG)测试:N氛围下,称取2.7mg左右PA12T 盐,以10℃/min 的升温速率从室温加热到600℃。
差示扫描量热法(DSC)测试:称取3mg左右的PA12T盐,N氛围下,以10℃/min的升温速率从室温升至300℃。
1.6.2 PA12T
FTIR表征:将PA12T样品干燥后研制成粉末,KBr压片,测定傅里叶红外变换光谱。
核磁共振氢谱(H NMR)测试:将PA12T 样品充分干燥,溶解在氘代三氟乙酸中,测定核磁氢谱。
黏度测试:称取PA12T 样品溶解在98%的浓硫酸中,溶液浓度为0.1g/mL,在25℃下用乌氏黏度计测试聚合物黏度。聚合物溶液和浓硫酸的流动时间分别记为和。相对黏度()、增比黏度()和特性黏数([])计算见式(1)~式(3)。

DSC 测试:称取3.2mg PA12T 样品,N氛围下从室温升至350℃左右,保温5min,后再降温,最后再升至350℃,升温和降温速率均为10℃/min。
TG 测试:N氛围下,称取2.7mg PA12T 样品,以10℃/min的升温速率从25℃加热到600℃。
2 结果与讨论
2.1 PA12T盐的TG和DSC分析
图2 和 图3 分别 是PA12T 盐的TG 和DTG 曲线图。由图2 可以看出,从204℃开始PA12T 盐出现失重现象,至500℃失重完全,在失重完全之前的温度范围内,出现两个明显的失重台阶,第1个失重台阶主要是由PA12T 盐分解出的C12DA 挥发导致的。由图3 可以看出,温度高于200℃时,DSC基线开始出现轻微波动,在257℃开始明显吸热,273℃处出现吸热峰值(PA12T盐熔点)。PA12T盐为PTA和C12DA形成的有机晶体,随着温度升高,会出现PA12T 盐熔融、分解、C12DA 与PTA 的挥发和聚合反应以及缩合物的分解等过程。值得特别关注的是,PA12T 盐在温度为200℃左右时即开始出现分解失重现象,此时的温度远低于其熔点(273℃),该现象对PA12T 的聚合过程控制具有十分重要的价值。

图2 PA12T盐的TG和DTG曲线图
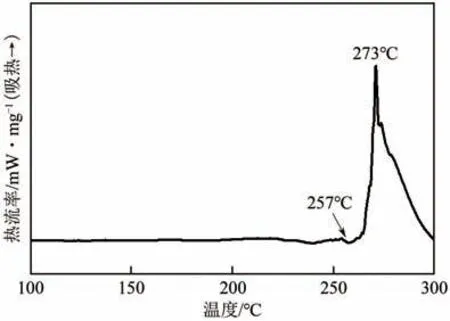
图3 PA12T盐的DSC曲线图
2.2 聚合反应条件对黏度的影响
2.2.1 升温速率控制
(1)水用量 由于聚酰胺的低介电损耗,几乎不吸收微波能量,本研究通过在反应体系中均匀分布少量水来吸收微波,达到反应体系快速均匀升温的目的。图4是水用量对聚合产物特性黏数的影响曲线。由图4 可以看出,当水的用量为0.17mL时,聚合产物PA12T 的特性黏数最高。低于该用量时,水用量的增加有利于聚合反应进行;高于该用量时,水用量的增加则会抑制聚合反应的进行。水是极性分子,在微波作用下其偶极子可以随微波电场方向的改变而快速运动,有利于快速升高温度,使反应物活化,促进聚合反应进行。但同时,水也是缩聚副产物,反应体系中水的存在会阻碍缩聚平衡反应向聚合方向进行,抑制分子量的增大。因此,水的用量是微波辅助聚合过程一个十分重要的影响因素。
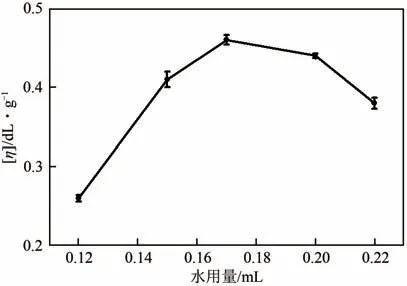
图4 PA12T的黏度与水用量的关系曲线
(2)聚合温度 图5是聚合温度与产物特性黏数的关系曲线。由图5可以看出,随着反应温度的升高,聚合产物PA12T 的特性黏数增大,当到达一定温度后,特性黏数随温度升高增大不明显。较高的温度有利于更多的反应分子被活化,使反应速率提高。在本反应条件下,从节能和聚合效率的角度考虑,较佳的聚合温度为210℃。需要指出的是,反应物PA12T 盐的熔点为273℃,聚合反应温度远低于其熔点,但聚合产物已不是PA12T 盐的粉末状,而呈现熔融结块,表明在反应过程中PA12T盐发生了熔融。

图5 PA12T的黏度与聚合温度的关系曲线
(3)保温时间 对于缩聚反应来说,在一定反应温度条件下,保温时间延长有利于聚合物特性黏数增大。图6是保温时间对聚合产物特性黏数的影响曲线。由图6 可以看出,随着保温时间的延长,特性黏数前期增加快,后期增加缓慢。当反应到达一定时间后,产物特性黏数增长缓慢。出现这种现象,认为主要是因为微波辅助聚合反应是在一个密闭反应器中进行,缩聚副产物水没有排出反应体系,当水的浓度达到一定程度后,会抑制平衡向聚合方向进行,使特性黏数增长缓慢。在本反应条件下,较佳的保温时间为60min。
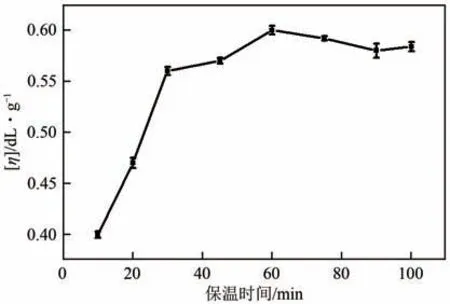
图6 PA12T的特性黏数与保温时间的关系
2.2.2 微波功率控制
微波功率决定微波的电场强度,对相同的化学反应体系而言,升温速率与微波功率成正增长关系。图7是微波功率与升到反应温度所需时间的关系曲线。由图7可以看出,当微波功率小于60W时,随着微波功率增大,升温速率快速增大;当微波功率大于60W 时,微波功率对升温速率影响不大。产生该现象的原因与微波穿透反应物程度有关。当微波进入物料后,介质吸收微波能并将其转变为热能,微波的场强和功率就不断地被衰减,衰减状态决定着微波对介质的穿透能力。当微波功率小于60W 时,随着功率增大,穿透能力增强,使升温速率增大;当功率达到60W 时,微波在反应物中的分布趋于均匀,功率对升温速率的影响减弱。

图7 微波功率与PA12T升温至聚合温度(210℃)所需时间的关系
图8 是特性黏数与微波功率的关系曲线。由图8可以看出,当微波功率小于60W时,产物的特性黏数随着微波功率增大而增大;当微波功率在60~100W 之间时,微波功率对产物特性黏数影响较小。在微波功率小于100W时,微波功率对产物特性黏数的影响规律与微波功率对升温速率的影响规律是一致的。但是当微波功率大于100W时,产物特性黏数随微波功率增大反而有所下降。由本文2.1 节可知,当温度大于200℃时,开始有C12DA挥发现象发生。随着微波功率增大,升温至聚合温度(210℃)所需时间缩短,未来得及与PTA 反应而挥发的C12DA 比例增大。当微波功率为150W、200W 和250W 时,反应结束取出产物时发现,聚合反应瓶壁上均有未反应的C12DA 液滴附着。表明在该条件下,C12DA 挥发现象十分明显,由于C12DA 的挥发,明显影响羧基和氨基的等摩尔比状态,导致产物特性黏数下降。
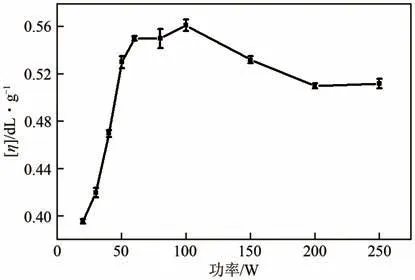
图8 PA12T的特性黏数与微波功率的关系
2.3 PA12T表征
2.3.1 FTIR分析
图9 是PA12T 盐和PA12T 的红外谱图。图9中,3322cm附近是—NH 伸缩振动的吸收峰,2922cm和2850cm附近是—CH—对称和不对称伸缩振动的吸收峰。1623cm附近是羰基伸缩振动峰(酰胺Ⅰ带),1539cm附近是N—H弯曲及C—N伸缩振动组合(酰胺Ⅱ带),1378cm附近是酰胺特征吸收带Ⅲ,862cm附近是苯环上—CH弯曲振动的吸收峰,722cm和639cm附近是苯环弯曲振动的吸收峰,这些特征峰表明酰胺键的存在。对比PA12T盐和PA12T聚合物的红外谱图,3000cm处是PA12T 盐中—NH+的宽而强的振动吸收峰,2205cm处—NH的倍频、合频吸收峰消失,出现了3059cm附近—NH和—CN的伸缩振动的组合倍频,2925cm处吸收峰面积显著增大,这些现象表明PA12T盐已经转变为PA12T。
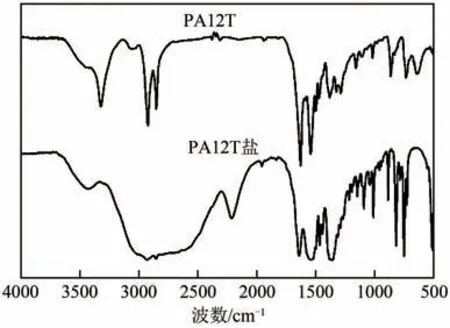
图9 PA12T的红外谱图
2.3.2H NMR分析
图10为PA12T的H NMR谱图。PA12T结构单元中含有4种化学环境不同的质子,各质子的化学位移()归属和质子峰的积分面积见表1。从表1中可以看出,聚合产物各质子峰的积分面积之比与PA12T 结构单元中质子数之比基本吻合,表明合成的聚合物是PA12T。

表1 PA12T的核磁氢谱分析

图10 PA12T核磁谱图
2.3.3 TG分析
图11和表2分别是微波辅助聚合和传统聚合最优反应条件下得到的PA12T的TG和DTG曲线以及对比分析数据。从图11和表2中可以看出,微波辅助聚合的PA12T 的初始分解温度(434℃)远高于传统聚合方法得到的产物(378℃),而最大分解速率温度和分解完全温度则基本相同。表明微波辅助聚合得到的产物中低分子量的分子所占比例较低,传统聚合产物中低分子量的分子所占比例较高。说明微波条件下的反应更为均匀,分子量分布较窄。PA12T是一种新型的半芳香尼龙,除了浓硫酸和三氟乙酸外,其他溶剂难以将其溶解,市售凝胶渗透色谱(GPC)尚不能用来快速测定其分子量和分子量分布。因此,利用TG 判断不同PA12T 产物分子量分布的相对宽窄不失为一个好的方法。由表2还可以发现,微波辅助聚合反应时间85min、传统聚合5h 时,微波聚合的时间虽短,但其产物的分子量却大于传统聚合产物的分子量。出现这种现象的原因是传统聚合方法升温过程中温度梯度的存在,造成产物的分子量分布比微波聚合产物宽。两种聚合方法的聚合反应在210℃达到平衡时,仍不能从根本上改变传统聚合方法分子量分布宽的现实情况,导致最终产物低分子量聚合物含量较高,测得的平均分子量也较小。研究结果充分说明,微波辅助聚合在大幅度提高聚合效率的同时,由于温度场均匀可以有效降低聚合物分子量分布。
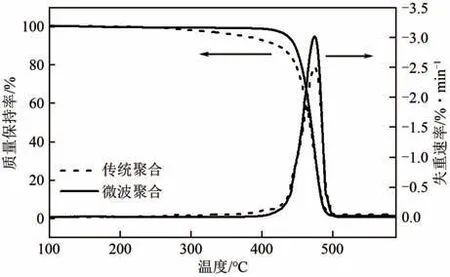
图11 PA12T的TG和DTG曲线
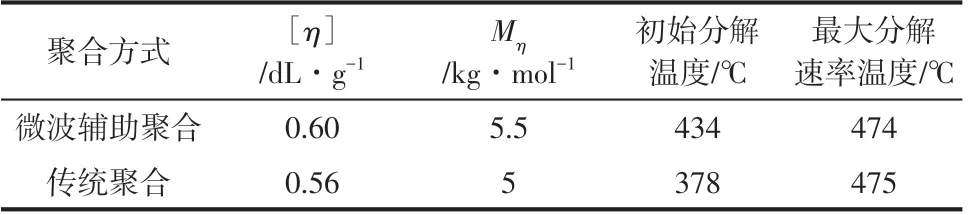
表2 PA12T的TG分析结果
2.3.4 DSC分析
图12 是PA12T 样品的升温和降温DSC 曲线。由图12 可以看出,微波辅助聚合与传统聚合样品的升温曲线均出现双重熔融吸热峰,其中低温峰一般情况下被认为是由不完善结晶熔融导致的。微波辅助聚合样品的熔点为300℃,传统聚合样品的熔点为287℃,微波辅助聚合样品的熔点比传统聚合样品的熔点高13℃。微波辅助聚合与传统聚合样品降温曲线的峰形出现了更大的差异,微波辅助聚合样品的结晶峰峰形清晰、宽度较窄,而传统聚合样品的结晶峰峰形不规则且宽度大。高分子物理理论指出,当高聚物的分子量小于临界分子量时,聚合物熔点随着分子量增大而上升。微波辅助聚合样品的特性黏数大于传统聚合样品,表明微波辅助聚合样品的分子量较大,熔点也较高,符合高聚物熔点与分子量的一般规律。微波辅助聚合与传统聚合样品降温曲线出现的较大差异,可以认为是由于微波辅助聚合样品中低分子量聚合物含量很低而传统聚合样品中低分子量聚合物含量高造成的,进一步支持了TG 分析认为的微波辅助聚合样品的分子量分布较窄的结论。

图12 PA12T的DSC曲线图
3 结论
本文通过微波辅助聚合方法在密闭反应器内合成了半芳香尼龙PA12T 预聚物。与传统聚合方法相比,聚合反应周期大幅度缩短,聚合反应更为均匀,分子量分布较窄,为后续固相后聚合反应得到高质量PA12T 产物奠定了极为有利的基础。受微波反应器结构所限,本研究在反应过程中无法排出聚合反应副产物水,尚不能得到高分子量聚合产物。但毫无疑问,本研究展现了微波辅助聚合高效且均匀的巨大优势,为继续深入进行半芳香尼龙高效聚合方法研究及微波反应装置设计和改进,提供了一个有价值的参考。
