非酒精性脂肪性肝病相关肝细胞癌的研究进展
2022-02-11辜雪莲李俊峰毛小荣
辜雪莲, 李俊峰, 毛小荣
1 兰州大学第一临床医学院, 兰州 730000; 2 兰州大学第一医院 感染科, 兰州 730000
肝细胞癌(HCC)占原发性肝癌的90%,是全球癌症相关死亡第三大原因[1]。据国际癌症研究机构数据,2020年全球约有90万新发HCC,83万死亡HCC病例,其中我国新发及死亡HCC病例数近总数的1/2[2]。目前病毒性肝炎仍是我国HCC发生最常见危险因素,但随着核苷(酸)类似物及疫苗的应用,其发病趋于稳定。近年来随着国人饮食及生活方式的改变,非酒精性脂肪性肝病(NAFLD)已取代病毒性肝炎成为我国最常见的慢性肝病,预计到2030年我国NAFLD病患数将达3.14亿例[3]。而NAFLD疾病过程并非完全良性,据2015年全球健康评估数据,我国NAFLD相关HCC死亡人数占同期HCC死亡总数的10.5%[3]。
NAFLD发展为HCC的具体机制仍不明确。目前普遍认为肝硬化是NAFLD发展为HCC的主要危险因素,欧洲肝病学会(EASL)、美国肝病学会(AASLD)、亚太肝病学会(APASL)指南仅建议对NAFLD相关的肝硬化患者进行HCC常规筛查[4-6]。然而荟萃研究分析结果表明,约38%NAFLD患者在无肝硬化背景下发生HCC[7],且与病毒性肝炎相比,NAFLD相关HCC患者诊断后生存时间更短[8],这提示NAFLD相关HCC的发病机制或许不同于病毒性肝炎。研究表明慢性炎症、脂毒性、胰岛素抵抗(IR)以及肠道菌群失调参与NAFLD相关HCC病理过程,且针对以上机制的治疗方式如靶向肠道菌群、IR、氧化应激等对防治NAFLD相关HCC亦具疗效。
1 NAFLD相关HCC流行病学概况
NAFLD包括肝脂肪变、非酒精性脂肪性肝炎 (NASH)、肝纤维化、肝硬化、终末期肝病和HCC。据估计全球约1/4的人口患有NAFLD,预计未来10年NASH的发病率将增加56%[9]。NASH已成为欧美等西方国家HCC发生的重要基础肝病。据美国器官移植科学登记处数据,2016年与2002年相比,近10年间与NASH相关HCC肝移植等候者增加近11倍,且NAFLD相关HCC发病率以每年9%的速度增长[10-11]。其他西方国家同样观察到这一趋势,在法国1995年—2014年NAFLD相关HCC患病率从2.6%增至19.5%[12]。尽管我国NAFLD相关HCC的具体发病率尚缺乏,但NAFLD发病率却迅速增加。据流行病学研究[13],2008年—2018年我国NAFLD发病率从18%增至29%,且我国患者发病中位年龄最小,这在未来将对我国医疗卫生系统造成巨大负担。
2 NAFLD相关HCC发生机制
NAFLD相关HCC发生机制主要从以下几个方面进行阐述,具体过程见图1。
2.1 脂肪组织炎症 脂肪组织储存过量能量,并参与肥胖患者体内慢性炎症建立。脂肪细胞和巨噬细胞分泌的炎症因子,如肿瘤坏死因子(TNFα),白介素(IL-1、IL-6)等在肥胖情况下上调[14]。TNFα、IL-6激活非脂肪组织中JAK/STAT3促瘤信号,导致肝细胞增殖凋亡失控而促进HCC发生。TNFα激活核因子κB(NF-κB)和c-Jun氨基末端激酶(JNK)放大炎症信号,活化肝星状细胞(HSC)并将其转化为促瘢痕形成的肝脏肌成纤维细胞(HMF)从而导致肝纤维化,HMF和招募的免疫细胞构成促癌微环境增强NF-κB信号并促进HCC形成[15]。此外,肥胖导致的肝脏氧化环境可使T淋巴细胞蛋白酪氨酸磷酸酶(TCPTP)失活,增加STAT1和STAT3信号传导。STAT1信号募集T淋巴细胞参与NASH及肝纤维化进展,但与HCC的发生无关,STAT3信号则参与HCC的形成[16]。这可能是NAFLD相比于其他慢性肝脏疾病更容易在无肝硬化背景下发展为HCC的原因。
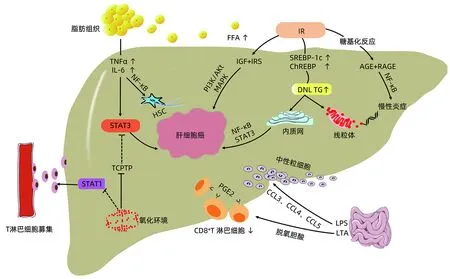
图1 NAFLD相关HCC发病机制
2.2 氧化应激及内质网应激(ERS) 肝细胞异常脂质蓄积是NAFLD的重要病理特征。毒性脂质如神经酰胺可抑制β-氧化、增加活性氧(ROS)产生,导致线粒体功能损害。二酰基甘油和神经酰胺还激活NF-κB通路致炎症信号进一步放大[17]。肝细胞脂质过度蓄积导致内质网(ER)功能紊乱,在生理条件下ER可通过激活未折叠蛋白反应(UPR)以适应这一压力[18]。尽管短暂的UPR可防止肝细胞脂肪变性,然而高脂饮食、毒性脂质如棕榈酸等可导致慢性ERS,这会加重肝脂肪变甚至诱发HCC发生。如前所述肝细胞NF-κB和STAT3激活是驱动NAFLD相关HCC的关键。ERS支路IRE1α磷酸化IkB蛋白激活NF-κB途径,促进TNF和IL-6等促瘤因子产生。在人类HCC的肿瘤组织中也发现较高的IRE1α与STAT3磷酸化相关[19]。此外,ERS导致胞质钙超载诱发线粒体功能障碍,产生大量ROS,导致DNA结构损伤而诱发HCC。
2.3 肠道菌群 肝脏与肠道通过门静脉及胆道系统相关联,肠-肝轴在NAFLD相关HCC发展中起重要作用。肠道菌群失调通过增加肠道通透性、影响胆汁酸代谢及抑制肝内免疫,而促进NASH甚至HCC[20]。在NAFLD患者中观察到肠道细菌过度生长及肠道通透性增加,这使脂多糖、脂磷酸壁(LPS、LTA)等病原体相关分子模式与肝内细胞接触。LPS与Toll样受体4(TLR4)结合,激活NF-κB信号通路诱发慢性炎症状态,营造利于肿瘤发生的微环境,诱导HSC及单核细胞释放CCL3、CCL4和CCL5等趋化因子,募集中性粒细胞至肝脏,加剧纤维化进程[21]。肠道菌群失调导致胆汁酸代谢失衡,使脱氧胆酸肠肝循环增加。LTA与脱氧胆酸共同作用于TLR2诱导HSC表达PGE2等SASP因子,PGE2与免疫细胞上的EP4受体结合导致CD8+T淋巴细胞数量减少而抑制抗肿瘤免疫[22]。此外产酒菌增加导致内源性乙醇生成,触发ROS生成导致脂质过氧化、慢性炎症和肝细胞死亡使单纯性脂肪变向HCC发展[23]。
2.4 胰岛素抵抗(IR) IR是NAFLD的重要特征,NAFLD患者循环中游离脂肪酸(FFA)参与IR的发生[24]。IR和葡萄糖可分别激活固醇调节元件结合蛋白1c(SREBP-1c)和碳水化合物反应元件结合蛋白(ChREBP),导致肝脏新生脂肪(DNL)合成增加而加剧肝脂肪变[24-25]。胰岛素样生长因子(IGF)表达增加与IR相伴随。IGF对肝细胞发挥生长因子样活性,刺激细胞增殖、抑制细胞凋亡增加肝细胞癌变风险。胰岛素受体(IRS)与胰岛素或IGF结合,激活P13K/Akt和MAPK,促进HCC发生[26]。高糖不仅为肿瘤细胞能量代谢提供底物,持续的高血糖导致糖基化反应生成晚期糖基化终产物(AGE), AGE与晚期糖化终产物受体(RAGE)结合,激活NF-κB和炎症信号级联并产生ROS,诱导HCC发生[27]。最近一项研究[28]发现,高脂肪饮食诱导尚未发生HCC小鼠肝细胞产生类Warbug效应,这表明高脂诱导的葡萄糖代谢重构可能在肝细胞癌变进程中起重要作用。
3 针对发病机制的防治进展
3.1 靶向慢性炎症 肥胖诱导慢性炎症是NAFLD相关HCC发生的重要环节,合理的饮食及运动可减轻NAFLD患者体质量并改善慢性炎症状态[29]。NIH-AARP饮食和健康研究队列表明,持续的体力活动与较低的HCC风险相关,保持每周长达4 h的运动可使HCC的风险降低30%(HR=0.74,95%CI:0.57~0.96)[30]。流行病学研究[31]表明,较高蔬菜摄入量可使HCC风险降低39%(95%CI:0.50~0.75),在人群中每增加100 g蔬菜摄入量,患肝癌的风险就降低4%(95%CI:0.95~0.97)。
此外,IL-6/STAT3信号是连接脂肪组织炎症及HCC的重要通路。在小鼠中已观察到IL-6或gp130缺陷表型HCC发病率降低,抑制JAK和STAT3均能抑制HCC细胞系增殖和原发性肝癌的生长,因此STAT3抑制可能是NAFLD相关HCC潜在治疗靶点[32]。C188-9是一种新型小分子STAT3抑制剂,与STAT3的SH2结构域结合阻止其酪氨酸磷酸化和同源二聚体化。在Jung等[33]研究中C188-9处理的Pten基因敲除小鼠比对照组小鼠表现出更少的肝脂肪变,且C188-9处理小鼠的平均肿瘤大小[(6.24±3.02) mm3vs (25.25±10.60) mm3]和平均肿瘤负担[(9.36±5.67) mm3vs (38.18±17.76) mm3]较对照组小鼠显著降低。近期的一项研究[34]表明,阿托伐他汀可通过减少IL-6分泌和 STAT3激活,使细胞周期停滞于G0/G1期,而降低异种HCC细胞移植模型小鼠HCC生长速度。因此,对IL-6/STAT3信号新药研发及老药物的新利用或是防治NAFLD相关HCC的前进方向。
3.2 抗氧化应激 毒性脂质介导ROS积累促进NAFLD相关HCC发生。姜黄素是从姜黄中提取的天然多酚化合物,具有抗氧化、抗炎和抗癌作用。姜黄素通过下调NF-κB,抑制TNFα、IL-1和IL-6、环氧合酶2等,缓解蛋氨酸和胆碱缺乏诱导的小鼠内质网及线粒体功能障碍,抑制NAFLD进一步向NASH发展[35]。核因子E2相关因子2(Nrf2)是体内重要的抗氧化因子,姜黄素可上调Nrf2和谷胱甘肽,下调HCC中的ROS和HIF-1α表达,而抑制肿瘤基质血管生成及远处转移[36]。流行病学研究[37]表明,他汀类药物可降低2型糖尿病(T2MD)患者HCC的发生率,而胆固醇吸收抑制剂依泽替米贝通过p62依赖的Nrf2活化保护肝细胞免受FFA诱导的细胞凋亡,从而改善NAFLD并减轻氧化应激。此外,依泽替米贝还可抑制高胆固醇血症介导的血管生成,而抑制高脂饮食小鼠HCC生长[38]。
3.3 调节肠道菌群 肠道菌群紊乱导致的慢性肝脏炎症介导NAFLD相关HCC发生。肠道微生态制剂诸如益生菌等可丰富肠道微生物组多样性、改善肠道通透性,使NAFLD患者肝脏脂肪蓄积及炎症得到缓解,免受其进一步向NASH及HCC发展[20]。Prohep是由鼠李糖乳杆菌GG、大肠杆菌Nissle 1917和热灭活的VSL#3等比例复合而成的益生菌制剂。Li等[39]将小鼠分为肝脏肿瘤接种前1周(ProPre)或当天(ProTreat)饲喂Prohep、顺铂治疗及空白对照组,实验结果表明,与空白对照组相比Prohep治疗可显著降低小鼠肿瘤大小,且ProPre与顺铂治疗组相比肿瘤重量无明显差异。此外,ProPre组肿瘤重量明显比ProTreat组小,这提示早期喂养益生菌制剂可以带来更好的抗肿瘤效果。肠道来源的LPS激活TLR4导致慢性炎症推动NAFLD相关HCC的发展,已观察到抗生素如利福昔明可降低门静脉LPS水平而改善NASH小鼠肝纤维化[40]。此外,有学者提出抑制LPS受体TLR4可能是NAFLD相关HCC的另一治疗途径,尽管多黏菌素B及内毒素拮抗肽可能通过阻断TLR4而产生抗HCC效应,然而目前尚缺乏临床研究证实,且长期抑制TLR4可能导致免疫抑制而产生负面影响[41]。
3.4 改善IR mTOR信号网络在癌细胞代谢和增殖中起关键作用,二甲双胍等药物可减少循环中的胰岛素和IGF-1可能与其抗癌作用相关。二甲双胍可降低T2MD患者NAFLD进展为HCC的风险,在T2MD患者中二甲双胍使用组比对照组HCC发病风险降低(HR=0.25,95%CI:0.11~0.58)并且其总病死率及肝移植风险也降低(HR=0.42,95%CI:0.24~0.74)[42]。此外,在CCl4诱导的小鼠HCC模型中发现二甲双胍可显著降低实验组小鼠血清中AST、ALT水平,抑制小鼠HSC活化并减少肝细胞中脂质积聚及早期纤维化肝脏中瘤结节的形成[43]。
过氧化物酶体增殖剂激活受体(PPAR)由PPARα、PPARβ/δ和PPARγ组成,参与体内能量平衡和炎症相关过程调节。PPAR是开发NAFLD药物最有潜力的靶标之一。PPARα(贝特类)和PPARγ(噻唑烷二酮类)以及Saroglitazar、elafibranor等PPARα/δ双重激动剂,可不同程度地改善胰岛素敏感性,减轻脂质积累、炎症趋化因子分泌、降低促纤维化基因表达[44]。EASL和AASLD推荐吡格列酮用于T2MD患者活检证实的NASH[44]。此外,吡格列酮可通过阻断RAGE信号,导致NF-κB、CyclinD1的表达下降,从而有效抑制HCC的增殖和侵袭[45]。
4 小结与展望
随着对NAFLD病程进展的理解,NAFLD不再被认为是单纯的肝脂肪性变,而是可进展为NASH、失代偿期肝硬化甚至HCC的动态疾病。近年随着肥胖、T2MD、代谢综合征等疾病发病率的增加,NAFLD病患数在世界范围内呈上升趋势,与之相关的HCC亦对全球公共卫生系统造成不小的压力。目前NAFLD相关HCC的发病机制研究仍处于探索阶段,尚缺乏针对具体靶点的防治药物,且NAFLD患者HCC筛查现状也不尽人意。因此,在实际临床工作中患者及医生有必要更加重视相关危险因素的管理控制,以便于合理用药指导并帮助NAFLD患者取得更好预后。
利益冲突声明:所有作者均声明不存在利益冲突。
作者贡献声明:辜雪莲负责撰写论文;李俊峰修改论文;毛小荣负责拟定写作思路,指导撰写文章并最后定稿。
