全基因组水平蒺藜苜蓿反转录转座子IRAP分子标记开发及应用
2022-02-10尹晓凡魏娜郑淑文刘文献
尹晓凡,魏娜,郑淑文,刘文献
(兰州大学草地农业生态系统国家重点实验室,兰州大学农业农村部草牧业创新重点实验室,兰州大学草地农业教育部工程研究中心,兰州大学草地农业科技学院,甘肃 兰州 730020)
遗传多样性是育种计划的基础,全面了解植物物种的种群结构和遗传多样性谱系是其保护、管理和利用植物遗传资源的前提,而深入了解优良育种材料的种质多样性和相互关系是选择优良亲本组合和增加作物育种杂种优势的先决条件[1]。在传统育种中,形态学和表型标记已广泛用于遗传变异的分析,但这些方法仅限于改变表型和高度遗传性状方面,且存在较多的局限性和不稳定性(例如受限于土地面积和育种时间,以及不稳定的环境条件等)[2]。随着分子生物学技术的发展,各种DNA分子标记技术已被广泛使用。19世纪70年代,第一个基于DNA的分子标记技术——限制性片段长度多态性(restriction fragment length polymorphism,RFLP)被开发。随后,科学家相继开发了各种分子标记方法,例如随机扩增多态性DNA(random amplified polymorphic DNA,RAPD)、简 单 序 列 重 复(simple sequence repeat,SSR)、扩 增 片 段 长 度 多 态 性(amplified fragment length polymorphism,AFLP)、表达序列标签(expressed sequence tag,EST)和单核苷酸多态性(single nucleotide polymorphism,SNP)。分子标记主要是基于DNA序列的变异性或多态性,在基础和应用遗传研究(例如遗传图的构建以及基因或定量性状基因座的绘制)和育种(例如标记辅助选择和基因组选择)中发挥着重要作用[3]。近年来基于DNA的分子标记能够克服众多与表型有关的局限性,并能够在单位成本和时间效益内表现出较高的效率和优势,已成为构建种群、种质及物种之间DNA指纹图谱和遗传分析的桥梁[4]。
反转录转座子是一类在植物基因组中广泛存在的DNA序列,是植物基因组中最丰富的移动元素,几乎存在于所有高等植物中。其中,长末端重复序列(long terminal repeated sequence,LTR)反转录转座子是几乎所有真核基因组的主要组成部分,可占整个基因组的40%~90%[5]。反转录转座子包括长末端重复转座子(LTR—RT)和非长末端重复转座子(non-LTR)。LTR—RT根据蛋白质结构域分为Gypsy和Copia家族[6]。基于已开发的几种反转录转座子的分子标记技术,包括序列特异性扩增多态性(sequence-specific amplified polymorphism,SSAP)、反转录转座子扩增多态性(inter-retrotransposon amplified polymorphism,IRAP)和反转座子微卫星扩增(retrotransposon-microsatellite amplified polymorphism,REMAP),基于反转录转座子的插入多态性(retrotransposonbased insertion polymorphism,RBIP)和引物间结合位点多态性(inter-primer binding site polymorphism,iPBS)。IRAP标记最初由Kalendar等[7]开发,旨在识别不同的大麦(Hordeum vulgare)品种,IRAP标记使用从反转录转座子LTR序列设计的引物,对两个附近的反转录转座子之间的DNA序列进行PCR扩增,近几年该类型引物已广泛用于研究遗传多样性,遗传关系和评价种质资源。与其他标记相比,由于其较长的末端重复序列,在真核生物的基因组各个染色体中广泛分布,具有拷贝数多、特异性高、通用性强、信息丰富、可转移性等特征,可用于开发和应用各种分子标记[8]。由于这些特征,反转录转座子已成为近些年来研究植物遗传多样性的首选标记,并已被广泛用于各种植物遗传多样性和多态性分析,例如,利用IRAP分子标记技术分析了59份烟草(Nicotiana tabacum)品种间遗传差异,根据烟草反转录转座子Tnt1、Tto1、Tnd21序列设计的5个引物扩增出151个条带,其中多态性带145条,多态性比率高达96.03%[9];PDR1是豌豆(Pisum sativum)Ty1-copia家族反转录转座子,SSAP引物基于PDR1的多嘌呤域设计而成,用于区分豌豆15个品种和56份品系并构建遗传图谱[10];Holasou等[11]利用IRAP和REMAP标记技术评估伊朗49个小麦(Triticum aestivum)品种,通过9个IRAP引物和20个REMAP引物分别扩增出90和126个基因座,多态性比率分别为81.78%和86.40%。大量研究表明,同源性LTR—RTN植物科属之间的反转录转座子序列可用于整个物种[12]。
蒺藜苜蓿(Medicago truncatula)作为豆科模式植物是继拟南芥(Arabidopsisthaliana)、水稻(Oryza sativa)之后第3个完成全基因组测序的植物。蒺藜苜蓿与大多数豆科植物[紫花苜蓿(Medicago sativa)、大豆(Glycine max)、豌豆、三叶草(Trifolium hybridum)等]的遗传关系较近,并具有良好的共线性,尤其与紫花苜蓿具有很高的同源性。因而从蒺藜苜蓿获得的信息可以用于其他豆科植物,这对于挖掘豆科植物遗传资源,促进豆科作物和苜蓿等牧草的育种具有重要意义[13]。紫花苜蓿是最重要和广泛种植的豆类饲料作物,具有较高的营养质量和经济价值,其优异的营养价值使其非常适合奶牛和牲畜生产[14—15]。苜蓿不仅具有作为牲畜饲料的价值,而且在减少侵蚀和养分流失,增强土壤固碳和增加氮肥方面起着重要作用[16]。苜蓿杂种优势已被证明是提高牧草产量和其他重要性状的一种方法[17]。由于最早发展起来的形态学标记数量有限,表型选择费时费力,且易受外界环境及植物生长发育阶段等因素的影响,不利于在遗传、育种等研究过程中应用。然而分子标记的开发和运用,其拥有庞大的标记数量,且不受发育阶段及外界环境的干扰,因而在遗传学研究领域具有很高的利用价值。在模型物种(蒺藜苜蓿)中发现基因和确定基因功能相对容易,通过比较基因组学将遗传信息从蒺藜苜蓿扩展应用到其近缘物种已成为一种重要策略[18]。因此,本研究首次在蒺藜苜蓿的全基因组水平上基于LTR信息设计IRAP标记引物用于40份紫花苜蓿种质,评估鉴定不同紫花苜蓿品种的遗传差异性,对候选IRAP标记进行了大规模搜索和开发,以评估其潜力。
1 材料与方法
1.1 试验材料
试验于2018年4—9月进行。本研究共涉及40份紫花苜蓿种质资源用于苜蓿遗传多样性分析(表1)。其中20份国外品种由美国农业部国家植物种质系统提供,20份国内品种由农业农村部国家畜牧总站和国家种质资源库提供。上述测试材料于2018年4月种植在兰州大学临泽实验站。

表1 40份紫花苜蓿种质类型及来源Table 1 40 alfalfa germplasm types and sources
1.2 DNA提取和检测
每份苜蓿品种采用30个单株的新鲜叶片混合取样[19],使用CTAB法提取苜蓿基因组总DNA(gDNA)[20],通过1%琼脂糖凝胶电泳检测质量。用Nanodrop分光光度计和1%琼脂糖凝胶电泳(OD260/OD280≈1.8)测量DNA浓度和质量。将DNA样品稀释至25 ng·μL—1的工作溶液,并在4℃下保存直至使用。
1.3 引物设计与PCR扩增
运用LTR—FINDER[21]在全基因组水平分析鉴定蒺藜苜蓿LTR反转录转座子,利用Array Designer 4软件[22]设计开发IRAP引物标记。共设计出431个IRAP引物,进而根据染色体位置信息和IRAP上下游引物标记方法选择设计了69对引物组合,用于下一步实验(表2),并进行3次重复试验。引物由上海生工生物工程技术服务有限公司合成。
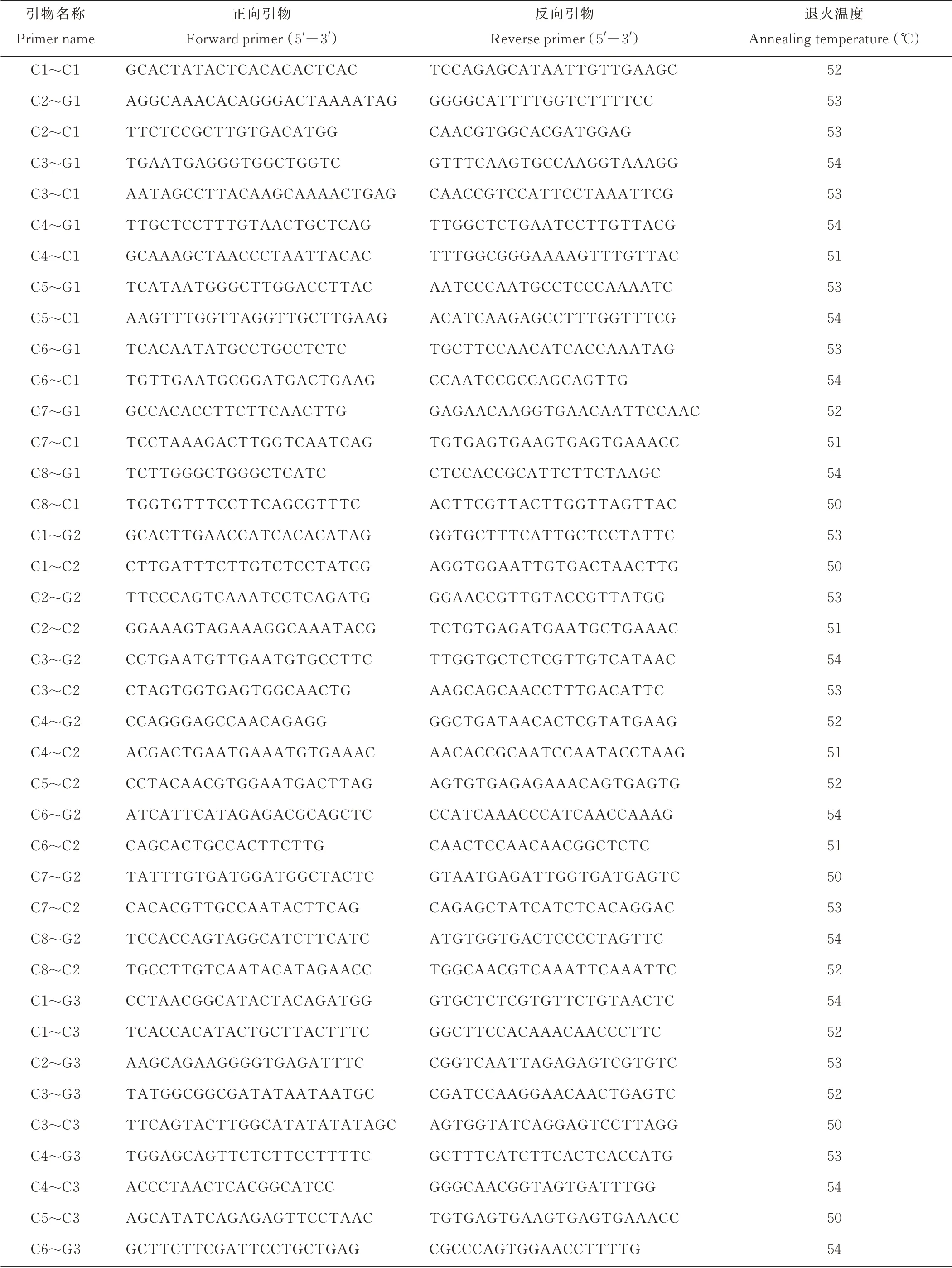
表2 引物信息及序列Table 2 Primer and sequences information applied in this study
PCR扩增在PTC-200热循环仪上进行:总体积为10μL,包含2×Taq PCR MasterMix 5μL,模板DNA 1μL,ddH2O 2μL,正向和反向引物1μL。PCR扩增程序如下:在94℃变性3 min;在94~51℃下退火30 s,在72℃下延伸30 s,9个循环;94℃变性30 s,50℃退火30 s,72℃延伸30 s,30个循环;72℃延伸7 min;储存于4℃冰箱。使用6%变性聚丙烯酰胺凝胶电泳(polyacryamide gel electrophoresis,PAGE)检测PCR扩增产物,将样品点样至2μL,并在210 V电泳110 min,并通过AgNO3溶液银染视化,并拍照保存电泳图谱。
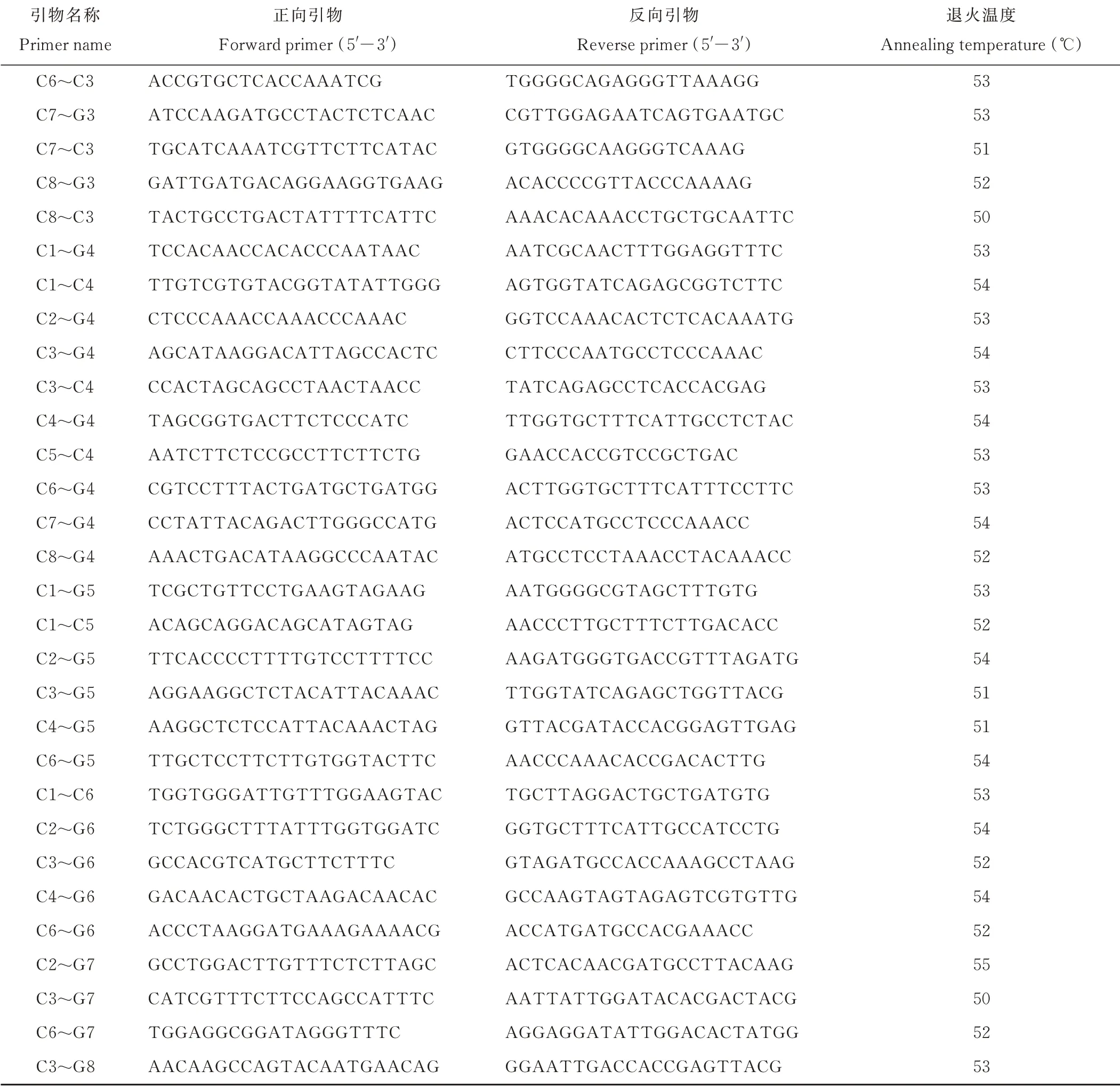
续表Continued Table
1.4 数据处理与分析
通过手动比较和软件校准对IRAP分子标记的扩增产物图谱进行统计分析。每个样品中所有可检测的扩增条带按照分子量编号读取,在相同的迁移位置有带(1)或缺失(0)进行评分,以建立“0,1”矩阵来确定遗传关系。使用软件NTSYSpc 2.1的Dice系数计算IRAP数据的基因型[23]。统计并计算总条带数(total bands,TB)、多态性条带(polymorphic bands,PB)、多态性条带比率(percentage of polymorphic bands,PPB)、预期杂合度(expected heterozygosity,He)和多态性信息含量(polymorphic information content,PIC)[24]。PIC通过PIC=1—∑(Pij)2的公式进行计算(Pij表示第i个等位基因和第j个等位基因的基因频率)[25]。使用算术平均值非加权组平均法(unweighted pair-group method with arithmetic means,UPGMA)产生相似系数矩阵,以此估计基因型之间的遗传相关性并生成树状图。使用Structure 2.3.1软件分析群体遗传结构[26]。估计最佳群体组群数(K),亚群的数量为1~11,最可能的K值由数据lnP(D)的对数似然性和STRUCTURE HARVESTER中的ΔK确定[27]。
2 结果与分析
2.1 IRAP标记的开发及分析
为了评估紫花苜蓿基因组中LTR多态性,将基于蒺藜苜蓿LTR反转录转座子设计的69对引物进行IRAP分子标记试验(表2)。根据多蛋白(polyprotein,POL)开放阅读框架和内部酶基因中所编码的蛋白质顺序,LTR反转录转座子又分为Ty1-copia和Ty3-gypsy两个超家族,即Copia和Gypsy家族。通过蒺藜苜蓿全基因组信息数据,对设计的431个IRAP标记引物按照LTR进行分类(表3)[28],分析已识别的LTR总数,检索序列的总长度(bp)及平均总长度(bp),平均配对分数以及用于分类和识别的LTR总数的百分比。其中,Gypsy家族占已鉴定LTR总数的24.40%(105),Copia家族占22.30%(96),未知的占53.40%(230)。Gypsy家族在LTR中的比例略大于Copia家族;总长度和平均长度也略大于Copia家族。

表3 基于蒺藜苜蓿全基因组序列设计引物分类信息Table 3 Information of designing primers based on the M.truncatula sequence
根据染色体位置信息对鉴定出的LTR进行染色体分布统计分析(图1),结果显示,其中未知(Unknown)主要分布在染色体1、2、3上;Gypsy家族主要分布在染色体1、2、3、4、6上,分别有11、15、22、14、16个,染色体5上分布最少,仅有2个;Copia家族主要分布在染色体1、3、5、8上,分别有17、18、11、14个,染色体2上分布最少,仅有6个。
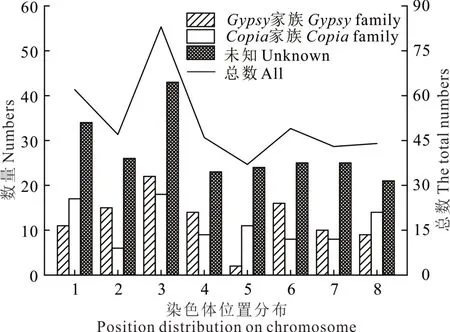
图1 IRAP引物染色体分布信息Fig.1 Chromosome distribution information of IRAP primers
2.2 IRAP标记的多态性分析
本研究开发设计出431个引物,并通过IRAP分子标记方法组合形成69对IRAP引物组合。PCR扩增筛选出37对具有高多态性和优良稳定性的引物组合,并对40份紫花苜蓿品种进行遗传多样性分析(表4)。
这些多态性引物组合可扩增出多态性高、清晰、重复性好的差异带型(图2)。由表4可知,37对多态性IRAP引物组合产生325条总条带和268条多态性条带,多态性条带比率为82.5%;每个引物组合的总条带数范围为3(C8~C1)至17(C1~G3,C2~C1),平 均 值 为8.8;多 态 性 条 带(PB)从2(C8~C1,C8~C2,C3~G4)到16(C2~C1,C1~G3),平 均 值 为7.2;多 态 性 条 带 比 率(PPB)为50%(C3~G4)至100%(C5~C4,C1~G5,C8~G2),平均值为79.9%;预期杂合度(He)的值为0.39(C8~C1)至0.89(C5~G1,C7~C2),平均值为0.73;多态性信息含量(PIC)的范围是0.34(C8~C1)至0.88(C5~G1,C7~C2),平均值为0.69,表明本研究的40个紫花苜蓿标准品种具有丰富的遗传多样性。根据上述数据和结果,引物组合C2~C1、C1~G3同时具有较大的TB、PB和PPB;引物组合C5~G1、C7~C2具有最大的He和PIC;引物组合C1~G3在PPB、He、PIC等方面优于大部分其他引物,He和PIC值分别为0.87和0.86。在苜蓿遗传多样性分析和品种鉴定中引物对C5~G1、C7~C2和C1~G3表现更好,潜力更大。
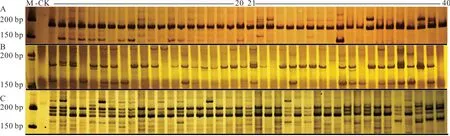
图2 部分IRAP引物在编号1~40的苜蓿品种中扩增结果Fig.2 IRAP amplification results of alfalfa varieties coded from 1 to 40 with part of primers
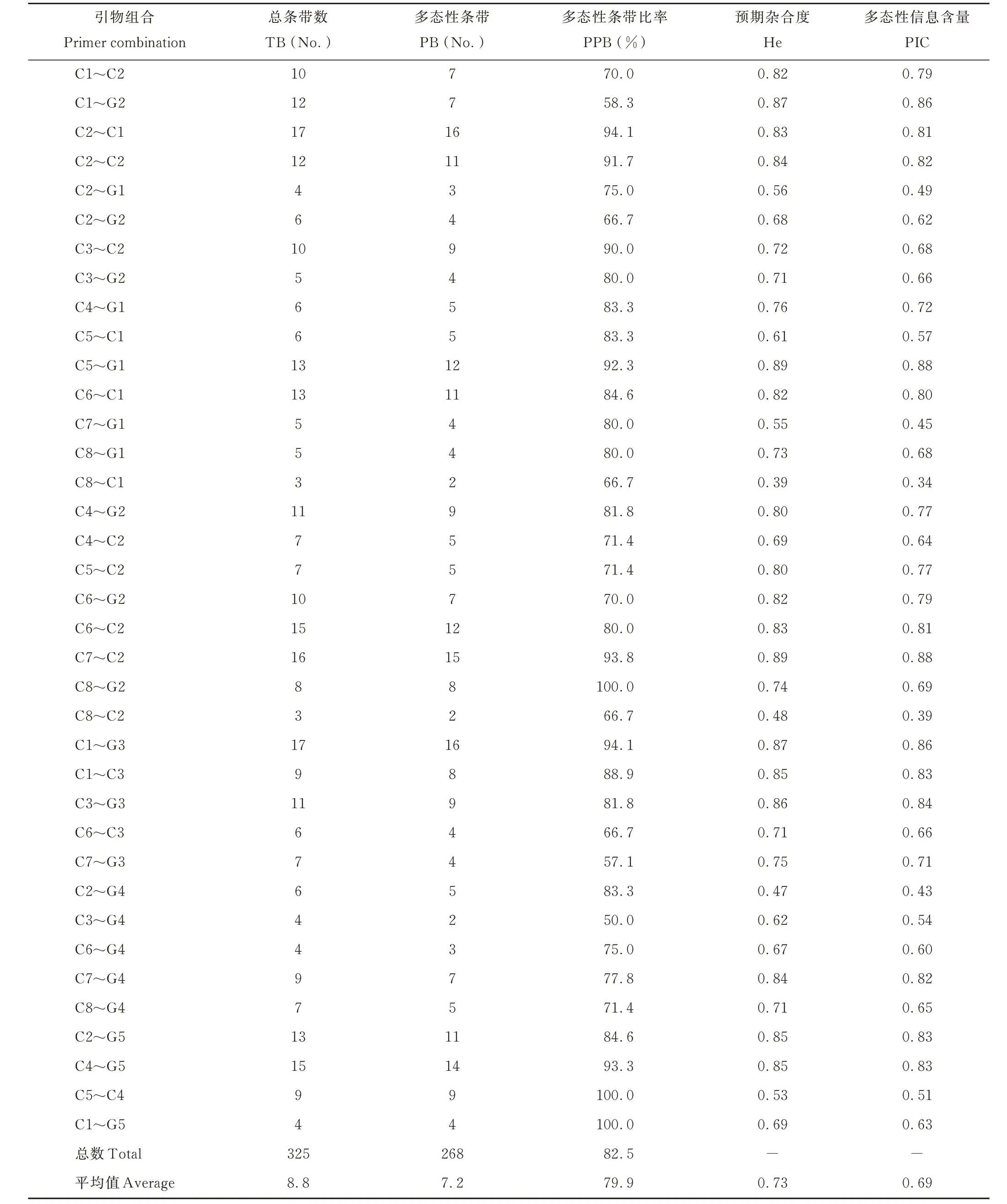
表4 37对多态性IRAP标记引物信息Table 4 Information of 37 pairs of polymorphic IRAP marker primers
采用37对核心多态性引物组合对40个品种进行分析,扩增出特征谱带,即仅用1个特征引物即可将品种区分开。在筛选出的引物组合中,每对多态性引物分别能扩增出2~16条多态性条带(表4)。不同的引物组合能够区分不同的苜蓿品种(1~7个),部分引物组合一次只可鉴定出一个品种,引物组合C2~C1能够扩增出16条多态性条带,可一次性区分7个品种。不同的引物对进行组合可鉴定出高于单个引物组合的更多品种,大大提高对不同品种的鉴别能力。利用37对多态性引物组合在40个紫花苜蓿品种的电泳带型对引物间扩增结果的相关性进行分析[29]。分析结果如图3所示,37对引物组合间,C3~G4与C6~G4之间相关性最高,相关系数达到0.95,代表两者之间扩增带型最相似,差异最小;C2~C1与C2~G2、C2~G4之间相关性最差,相关系数均为—0.31,表明C2~C1与两者之间带型差异最大;C2~C1与C4~G1、C5~C1、C6~C3之间相关性系数也达到—0.30,带型差异也很大。因此,根据各个引物组合间相关系数以及扩增结果相关性,可以进一步辅助优化不同品种间鉴定的引物选择和组合,即利用相关性较差的引物组合更加利于进行品种间的鉴定。

图3 IRAP引物扩增结果相关性分析Fig.3 The correlation analysis based on the IRAP productions
2.3 遗传相似系数聚类分析
根据37对IRAP多态性引物的扩增结果建立了1与0形式的数据矩阵,利用NTSYSpc 2.1软件录入各品种间遗传相似系数。通过UPGMA聚类分析,以遗传相似系数为基础构建了40个苜蓿品种的聚类分析图(图4)。根据聚类结果,相似系数以0.82为阈值可以将40个苜蓿品种分为4个主簇聚类(Ⅰ,Ⅱ,Ⅲ和Ⅳ)和两个亚簇聚类(Ⅱ-Ⅰ和Ⅱ-Ⅱ),所有品种均可区分。Ⅰ主簇具有8个种质;Ⅱ主簇有23个种质并分为两个亚簇,Ⅱ-Ⅰ子类包括全部来自中国的12个种质,Ⅱ-Ⅱ子类包含来自国外的11个种质[包括美国(4)、奥地利(2)、沙特阿拉伯(2)、英国(1)、波兰(1)、墨西哥(1)];Ⅲ主簇具有6个种质均来自国外[美国(3)、法国(2)、危地马拉(1)];Ⅳ主簇仅具有3个种质,均来自美国。从聚类分析图中可知,国内20个品种均聚集在Ⅰ主簇和Ⅱ-Ⅰ亚簇,国外20个品种聚集在Ⅱ-Ⅱ、Ⅲ、Ⅳ簇。聚类分析结果表明,品种间的遗传基础较为狭窄,遗传距离较接近,相似系数较高;地理起源相同的品种具有相似的遗传背景;品种CUF101、Trifecta、新疆大叶分别各自聚为一子类,表明品种间遗传背景较远,多样化程度较高。如选取同一类群的苜蓿品种(系)进行杂交选育,成功率较低,可能很难达到期望水平。但选取2个不同类群的苜蓿品种进行杂交选育,成功率较高,可能会大大缩短优良品种的选育进程。同时,本研究也表明IRAP分子标记方法适于紫花苜蓿种质资源的亲缘关系分析、鉴别及育种遗传距离的判定等相关研究。
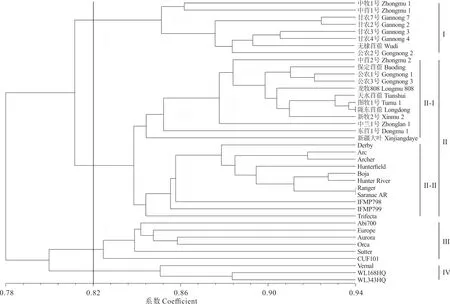
图4 苜蓿IRAP标记UPGMA聚类分析Fig.4 The UPGMA cluster analysis of IRAP marker in alfalfa
2.4 群体结构分析
STRUCTURE是一种类似于主成分分析(principal component analysis,PCA)和进化树的方法,使用分子标记的基因型信息对一组样本进行分类[30]。使用IRAP数据对40个苜蓿品种进行了结构分析。所有K值的簇数(K)为1到11,并且该操作重复20次。当K设置为3时,获得最高概率(图5A),最大ΔK出现在K=3处(图5B),之后ΔK值随K的增加而降低,并且当K>3时再未观察到ΔK的峰值(图5B)。ΔK为54.06,代表3个亚群(图5C),图5D显示了在K=3时的条形结构图。在K=3时,苜蓿品种大致分为国内品种和国外品种,结构分类图表明这些紫花苜蓿材料结构组成来源广泛,遗传背景复杂,亚类间有基因交流。蓝色组是最大的组,由国内品种(中国)组成;绿色和红色组主要由国外品种组成,UPGMA聚类结果与STRUCTURE分类结果基本一致。
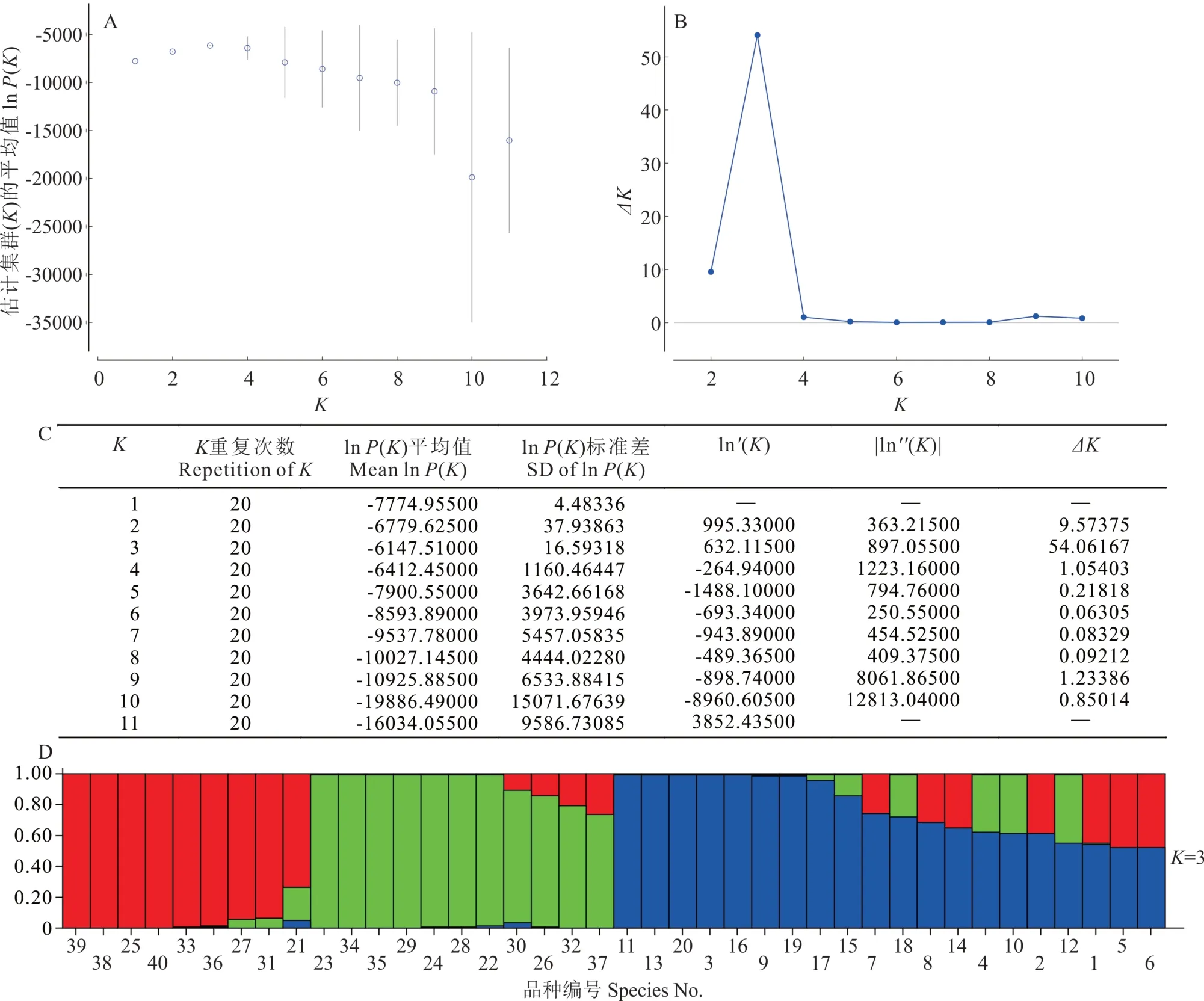
图5 苜蓿IRAP标记STRUCTURE分析Fig.5 STRUCTURE analysis of IRAP markers in alfalfa
3 讨论
IRAP标记作为LTR反转录转座子分子标记的一种,可检测在宿主基因组中的反转录转座子插入多态性,易于操作,不需要进行酶切及引物衔接子等繁琐过程,因此成为应用较广泛的LTR标记之一[31]。先前研究表明在马铃薯(Solanum tuberosum)全基因组LTR转座子分析中,平均长度为786 bp[32],本研究中蒺藜苜蓿全基因组水平上LTR序列信息中反转录转座子LTR的平均长度为2605 bp,远超过马铃薯中LTR平均长度,并且在各个染色体上均有分布。反转录转座子的大小随植物种类的不同和反转录转座子类群的差异而变化很大,反转录转座子两侧翼具有长末端重复序列(LTR),其长度从100 bp到5000 bp不等,在植物中拷贝数多,且散布于各染色体,非常有利于分子标记的开发。不同的物种具有不同的LTR—RT组成:在拟南芥中Gypsy与Copia元素的比例为1.5∶1[33—34];蒺藜苜蓿中Gypsy与Copia元素的比例为1.1∶1;低于玉米(Zea mays)(1.6∶1)和高粱(Sorghum bicolor)(3.7∶1)[35],但与大豆(1.4∶1)接近[33]。另外,研究表明,基于LTR—RT开发设计的引物可用于密切相关的属[6]。先前在豆科植物中运用的IRAP引物是基于早先在百脉根(Lotuscorniculatus)(LORE1、LORE2)和豌豆(Tps12a和Tps19)豆科植物RTN家族设计[33—34],但由于不同物种的遗传特异性,LTR反转录转座子分子标记的开发需要依赖物种全基因组序列信息,因此目前尚未有基于豆科植物LTR分子标记开发和应用的相关报道。本研究在蒺藜苜蓿全基因组水平上开发了431个LTR引物,进而用于评价与其近缘关系最为接近的紫花苜蓿种质资源。
品种的遗传多样性分析着眼于揭示品种的遗传关系和遗传基础,部分引物可以直接区分某些紫花苜蓿品种。本研究中,IRAP标记能扩增出多态性高、清晰、品种间有差异的带型。Branco等[36]利用22个IRAP标记在51份水稻种质中扩增出156条多态性条带;Holasou等[11]利用9个IRAP标记评价49份小麦种质扩增出74条多态性条带;Carvalho等[37]利用5个IRAP标记在48份小麦种质中扩增出103条多态性条带;Mandoulakani等[12]利用10个IRAP引物组合评价8个种群80个紫花苜蓿基因型,扩增出66条多态性条带,多态性条带比率为65.3%。本研究筛选出的37对多态性引物,相比于其他物种,IRAP标记成功扩增出更多的多态性条带(268)以及较高的平均多态性条带比率(79.9%)。在多态性引物扩增结果中只要一个品种之间的一个DNA基因座(片段)的基因型(带型)不同,就可以认为是不同的品种;筛选出核心引物,可以使用不同的DNA条带型以及相关性较低的引物组合来区分更多的品种。在本研究中引物的PIC平均值为0.69,其中有19个(51.35%)引物的PIC>0.7,18个(48.65%)引物的PIC值范围为0.34~0.69。研究表明当PIC值大于0.7可用于构建遗传图谱[38],表明基于LTR开发多态性IRAP分子标记在遗传多样性和遗传图谱分析中具有较大的潜力。
聚类分析指将物理或抽象对象的集合分组为由类似的对象组成的多个类的分析过程,它已成为牧草育种的必要手段之一。在IRAP聚类分析中,多态性引物扩增结果在同样的阈值条件下,能够在品种间更好地将全部供试材料分成4大主簇,以及更细更具体的类群,从而更容易区分国内外40份紫花苜蓿种质资源的遗传多样性。利用内含子长度多态性(intron-length polymorphic,ILP)分子标记对21份紫花苜蓿种质资源的分类研究中,甘农4号、公农2号与国外品种被分在同一主簇;甘农3号单独分在一主簇,无法将国内外品种具体区分[16]。而本研究开发出的IRAP标记多态性引物组合能够将国内外种质资源进行区分,同时也将国内各个品种进行了更加具体的分类。紫花苜蓿品种的STRUCTURE分析中显示出各个集群包括国内外品种的混合,造成结构混合的原因有很多:例如,所有这些基因型都包含一定程度的相同等位位点,基因交流程度,异花授粉或祖先的常见遗传成分。大部分苜蓿品种是通过表型轮回选择或大量自然选择栽培的综合品种,多个亲本参与杂交,导致部分繁殖品种之间的系统发育构成有着或多或少的关联。不同品种间遗传变异程度不一致,可为紫花苜蓿品种改良提供丰富、优良的亲本条件,该研究结果比经验性的直观分类结果更准确可靠。因此,从整体来看,IRAP分子标记得到的多态性条带数目更多、更稳定、多态性条带比率高,更易揭示不同品种的遗传差异,可将紫花苜蓿种质更科学、更系统地分类,在紫花苜蓿种质资源遗传多样性研究上有重要的应用价值。
4 结论
本研究通过蒺藜苜蓿全基因组信息设计IRAP引物用于紫花苜蓿品种鉴别和种质资源分析。以37对核心多态性引物组合构建了40份紫花苜蓿品种的遗传多样性分析和品种鉴别体系。从分子水平快速准确鉴别和分析苜蓿品种遗传多样性,扩增谱带在品种中具有丰富的多态性,能较好地将不同品种区分开,为合理利用紫花苜蓿种质资源的分类、品种识别、选配优良杂交组合培育新品种提供理论依据。
