靶向蛋白质降解技术研究进展*
2022-02-10张晓元张艳艳孙晓康张林军
张晓元 张艳艳 孙晓康 张林军 陈 勉 刘 飞
(山东省药学科学院,山东省生物药物重点实验室,山东省多糖类药物工程实验室,多糖类药物发酵与精制国家地方联合工程实验室,济南 250101)
蛋白激酶、离子通道、受体等人体蛋白是当前药物开发最常用的药物靶标。截至2015 年,美国食品药品监督管理局(FDA)批准的1 578 种药物分子靶点中667 种为人源蛋白,占总药物靶点的75%以上[1]。随着人类基因组成功测序及不断深度解析,构成人类基因组的蛋白质编码基因的实际数目确定为2万多[2],存在大量潜在药物靶点。传统靶点确认及干预策略包括使用小分子拮抗剂与靶蛋白特定位点结合来阻断或调节蛋白质功能,然而,细胞内超80%蛋白质如骨架蛋白、转录因子和非酶蛋白等尚未发现有效小分子拮抗剂[3]。同时,该种“占位驱动(occupancy driven)”作用模式需持续占据靶蛋白活性位点,要求小分子拮抗剂具有高亲和力、长半衰期且一般使用量较大以满足靶点饱和,导致毒副作用大且易产生耐药;或采用DNA敲除、CRISPR‑Cas9及RNA干扰(RNAi)等从DNA和/或mRNA水平上影响基因表达,进而引起细胞总蛋白质水平降低,通常需几小时甚至几天才能形成效果,细胞有足够的时间建立代偿机制或者产生继发效应。另外,翻译前水平间接调控蛋白质表达技术存在调控不可逆性、成药性差、易脱靶等共性问题,仍需进一步完善。
人体细胞内数以千计的蛋白质正不间断进行转录、翻译、传导、催化、合成等功能,同时胞内受损蛋白、错误折叠蛋白、外来蛋白等即时降解为小分子氨基酸再利用和维持细胞自我平衡。蛋白质的合成和降解一直处于动态平衡中,受复杂机制监控,具有重要生理意义,一旦发生异常会导致肿瘤、神经退行性疾病等。细胞内蛋白质降解大都采用泛素蛋白酶体系统(ubiquitin‑proteasome system,UPS)和自噬(autophagy)系统两个途径[4]。近年来,科研人员基于上述途径开发出许多直接靶向蛋白质降解技术,探究了蛋白质在自然环境中如何发挥作用。区别于传统“占位驱动”,基于“事件驱动(event driven)”作用模式的靶向蛋白质降解技术,直接靶向并催化目标蛋白质降解,无需与靶向蛋白质长时间和高强度结合,低浓度化合物即可达到靶蛋白高效降解,具有高活性、高选择性、靶向“不可成药”靶点等诸多优势。本文详细介绍了基于UPS、自噬及内体‑溶酶体等途径的蛋白质降解技术最新方法、作用机制等,以期为蛋白质靶点快速高效筛选及靶向性药物开发提供理论与技术支持。
1 基于UPS蛋白质降解技术
UPS 是细胞内80%以上蛋白质降解的主要途径,由泛素(ubiquitin,Ub)、泛素活化酶(ubiquitin‑activating enzyme,E1)、泛素缀合酶(ubiquitin‑conjugating enzyme,E2)、泛素‑蛋白质连接酶(ubiquitin‑protein ligase,E3)、蛋白酶体及其底物蛋白等构成。
其中,泛素是一种由76个氨基酸构成的多肽,可在E1、E2、E3 三类酶级联反应催化下,特异与底物蛋白形成牢固的共价键进行多聚泛素化修饰,包括K6、K11、K27、K29、K33、K48、K63 位的修饰,其中K48多聚泛素修饰蛋白质会被细胞内蛋白酶体识别并进行降解(图1),泛素链被蛋白酶体相关的去泛素化酶(deubiquitinating enzymes,DUBs)去除并循环利用,而蛋白质底物则被折叠和降解。在此基础上,科研工作者开发了deGradFP、PROTAC、Molecular glue、dTAG、AID、Trim‑Away等系列蛋白质降解技术。
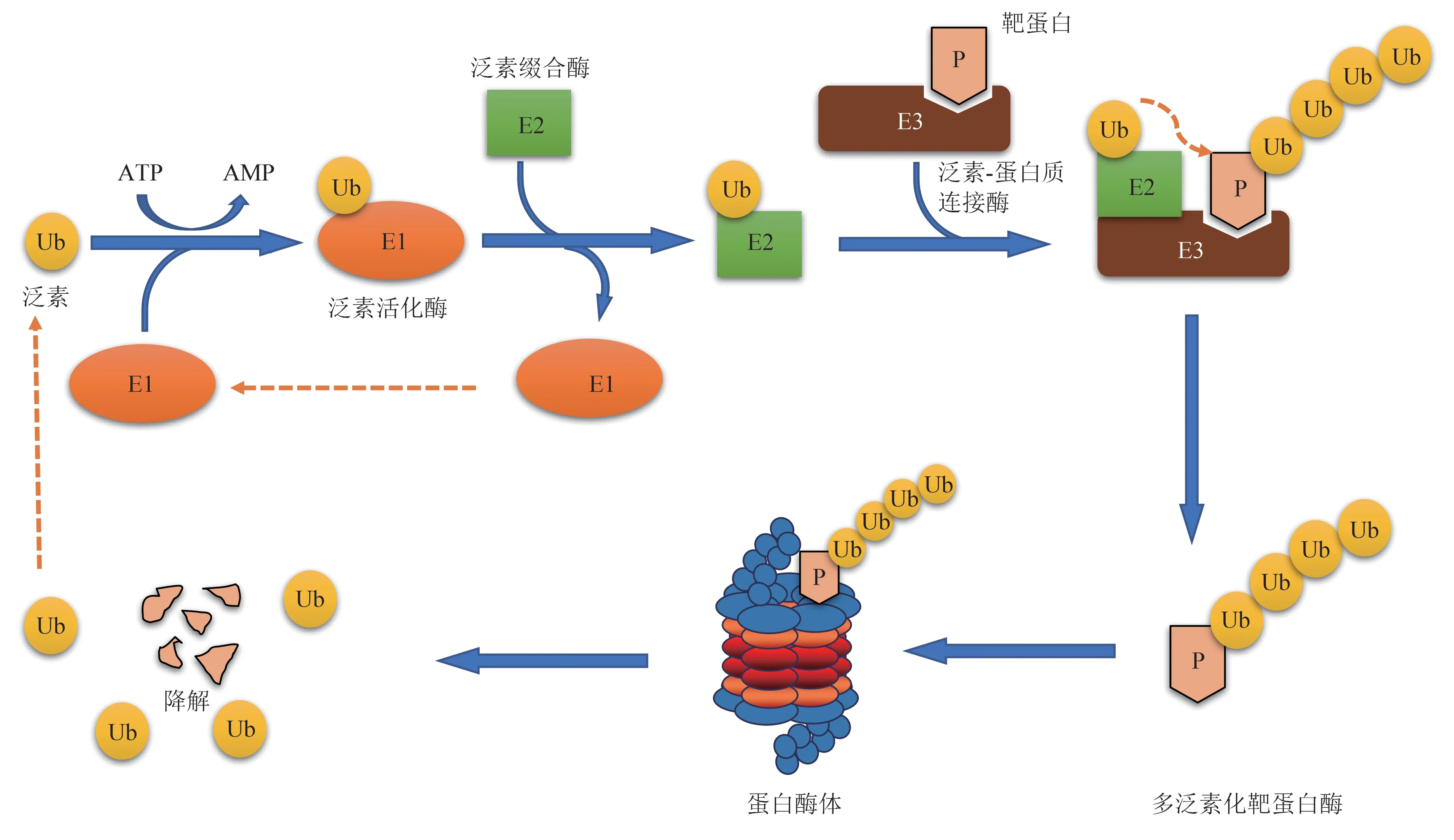
Fig.1 The process of protein ubiquitination and degradation图1 蛋白质泛素化降解的过程
1.1 deGradFP(degrade green fluorescent protein)
deGradFP技术由Caussinus等发明[5‑6],所采用的E3 为SCF 复合体(Skp1‑Cul1‑F‑box 蛋白),由S‑phase kinase associated protein 1(Skp1)、Cullin 1(Cul1)、Ring‑Box 1(Rbx1)/regulator of cullins 1(Roc1)和F‑box 四个亚基组成。其中Skp1、Cul1和Rbx1/Roc1 组成一般性骨架,F‑box 蛋白具有底物识别特性,决定SCF 复合物底物特异性。deGradFP技术依赖于融合到F‑box结构域的绿色荧光蛋白GFP 单结构域抗体片段(vhhGFP4),此融合蛋白可结合绿色荧光蛋白GFP、黄色荧光蛋白Venus、黄色荧光蛋白YFP 或增强黄色荧光蛋白EYFP 标记的功能蛋白,并直接介导K48 多聚泛素化修饰,最终被蛋白酶体降解(图2a)。荧光蛋白标记最大优点在于可快捷方便监控靶向蛋白质降解水平。Caussinus 团队利用该技术在果蝇体内靶向组蛋白H2A 变体(His2AV)降解实验表明,诱导表达28 min 后靶向蛋白质水平开始下降,151 min后残留不足10%,显示出优异降解效率。Shin等[7]将vhhGFP4 与E3 复合物的衔接蛋白SPOP 融合开发出新升级系统,引导更高效的核蛋白降解。deGradFP 技术已成功在烟草[8]、果蝇[6]、斑马鱼[9]和动物细胞系[10]中获得成功应用。
1.2 蛋白降解靶向嵌合体(proteolysis targeting chimera,PROTAC)
PROTAC是一种两头含有不同配体的杂合双功能化合物,分别为可结合E3 的配体和可与细胞内目标靶蛋白结合的配体,两配体间通过Linker 相连,形成“三体”聚合物——靶蛋白配体‑Linker‑E3配体[11]。其作用机制为靶蛋白配体将靶蛋白招募到E3 附近,实现靶蛋白泛素化标记,最后启动细胞泛素化水解过程被蛋白酶体降解,而PROTAC不被降解,可循环使用(图2b)。Raymond Deshaies和Craig Crews教授团队[11]于2001年发明PROTAC 技术。早期PROTAC 技术以多肽为配体,生物相容性高、细胞毒性低,但分子质量较高,细胞通透性及化学稳定性差,严重影响其成药性。故后续研究重点集中在开发小分子PROTAC 上,2008 年Crews 团队利用咪唑啉衍生物Nutlin 与E3鼠双微体2(MDM2)高亲和力特点,创造性与靶向雄激素受体(AR)的小分子SARM 相连,设计出用于降解AR的首个完全小分子形式PROTAC‑1,在HeLa 细胞中10 μmol/L PROTAC‑1 处理7 h 后,雄激素受体水平明显下降,证明了透膜小分子化PROTAC 开发的可行性,但诱导AR 降解的PROTAC‑1 浓度较高(微摩尔级别)。经近20 年研究,更多的可用于小分子PROTAC 设计的E3 被发现,包括Von Hippel‑Lindau(VHL)[12]、Cereblon(CRBN)[13]、凋亡蛋白抑制蛋白(IAPs)[14]、MDM2[15]和DCAF16[16]等。已有报道,靶蛋白包括核受体(ER、AR、RAR)、蛋白激酶(AKT、RIPK2、CDK9、BTK、TBK1、BCR‑ABL、CDK2/4/6/9、RIPK2、ALK、CK2、PI3K、ERK1/2)、蛋白质转录调控蛋白(BRD4、Sirt2、TRIM24、HDAC6)、调节蛋白(ERRα、FKBP12、TACC3)、神经退行性相关蛋白(Huntingtin、Tau、α‑synuclein)、胞外代谢酶(MetAP‑2、DHODH)、表观遗传相关蛋白(PCAF/GCN5)等30 余种蛋白质及其突变体成功利用PROTAC 技术进行降解[17‑23],涉及疾病包括肿瘤、类风湿、神经退行性疾病等,诱导降解PROTAC 浓度也不断降低,如BET 有效降解剂QCA570 在白血病细胞MV4‑11中半数抑制浓度IC50低至8.3 pmol/L[24],展现出良好应用前景。
浙江大学侯廷军教授团队[25]建立了集PROTAC 结构信息和实验数据于一体的开放式WEB 访问数据库PROTAC‑DB(http://cadd.zju.edu.cn/protacdb)。截至2021 年4 月17 日,该数据库囊括了2 258 个PROTAC、275 个弹头(靶向目标蛋白质的小分子)、65 个E3 配体和1 099 个Linker,并详细提供其化学结构、生物活性和理化性质,以及PROTAC的降解能力、结合亲和力和细胞活性。尽管已有丰富的PROTAC‑DB元件数据库信息,但PROTAC的发现、优化及理性设计仍十分困难,一直是研究重点之一。研究表明,当前PROTAC 蛋白降解的开发瓶颈是Linker长度构效关系(SAR),Linker 长度和化学成分会影响PROTAC 的结构刚性、疏水性和溶解性等[26]。随着结构生物学和生物信息学深入研究,Linker 长度SAR 及PROTAC设计已由经验性转向理性设计:一方面通过X射线晶体结构数据进行RosettaDock 模拟、分子动力学模拟等设计新的有效的PROTACs[27‑29],其 中,Ciulli 教授团队[27]于2017 年首次报道了PROTAC分子MZ1与VHL和靶蛋白受体Brd4BD2形成的稳定三元复合物共晶结构,为后续的理性PROTAC 药物分子设计奠定了基础;另一方面利用计算软件辅助理性设计,如Drummond 等[30‑31]利用MOE 软件开发出一系列生成和分析PROTAC 三元复合物协议、Zaidman等[32]开发PROsettaC软件,Bai等[33]开发基于Rosetta 的三元复合物建模协议等,最大限度地减少合成和生物评估分子的数量来提高筛选通量。
最近,科研人员针对PROTAC 进行系列创造策略来升级改造,包括开发出光可控PROTACs,如 pc‑PROTACs[34]、photoPROTACs[35]、PHOTACs[36]、opto‑PROTAC[37]等,可通过紫外线或可见光照射控制PROTAC 在特定组织中靶向降解目标蛋白质,避免可能出现的全身毒副作用;开发出可智能激活的靶向吲哚胺2,3‑双加氧酶(IDO)的半导体聚合物纳米 PROTAC(semiconducting polymer nano‑PROTAC,SPNpro),结合光学疗法和蛋白降解抗癌双重力量,在肿瘤中特异性激活PROTAC 来高效抑制小鼠肿瘤生长和转移,克服了当前PROTAC 分子潜在的脱靶副作用[38];开发出homo‑PROTACs,实现E3 分子双向多聚泛素化,介导E3自降解[39‑40]。
1.3 分子胶(molecular glue)
与PROTAC 杂合双功能小分子化合物不同,分子胶为单价小分子(通常分子质量<500 u),通过修饰重塑E3表面,诱导或增强E3和靶蛋白之间的相互作用,来实现识别并促进泛素化以降解靶蛋白(图2c)。目前较成熟的分子胶主要有两大类:一类为免疫调节药物(IMiDs)沙利度胺(Thalidomide)及其衍生物来那度胺(Lenalidomide)、泊马度胺(Pomalidomide)等,可与E3 CRBN 结合,招募含锌指(ZF)结构域因子的转录因子进行泛素化并被蛋白酶体降解,而IMiDs本身对降解转录因子没有可测量的结合亲和力。已报道降解靶点包括Ikaros 家族锌指蛋白1(IKZF1)、锌指蛋白3(IKZF3)、锌指蛋白91(ZFP91)、酪蛋白激酶1α(CK1α)及翻译终止因子GSPT1 等;另一类为芳基磺酰胺类,如吲地磺胺(Indisulam)、他西舒兰(Tasisulam)等可与E3 DCAF15 结合,招募剪接因子RNA 结合基序蛋白39(RBM39)进行泛素化并降解。上述两类分子胶的发现均具有偶然性,目前可用于靶向降解蛋白质的分子胶尚缺乏合理的筛选和设计策略,限制了其发现的效率和适用性。2020 年Mayor‑Ruiz 等[41]创新利用表型化学筛选法(phenotypic chemical screening)筛选新分子胶降解剂,发现一种新的RBM39 分子胶降解剂dCeMM1,验证了该方法可行性。Benjamin Ebert团队[42]则通过系统挖掘数据库及CRISPR‑Cas9筛选技术,关联分析578种肿瘤细胞系中499个E3组分的mRNA水平与4 518个临床和临床前小分子的药物敏感性,发现一种新分子胶细胞周期蛋白依赖激酶CDK 抑制剂CR8,CR8通过DDB1直接与CDK12‑cyclin K结合,并诱导与Cul4‑Rbx1‑DDB1 形成复合物,不依赖任何DCAF底物受体,使cyclin K发生泛素化并被蛋白酶体系统降解,进一步对CR8 结构特性分析发现CR8 分子胶的活性主要依赖于暴露在激酶表面的2‑吡啶部分,揭示合理修饰结合靶标的小分子的表面暴露区域可用于开发特定蛋白质靶标的分子胶降解剂。
1.4 dTAG(degradation TAG)
Nabet等[43]开发的快速、广谱dTAG蛋白降解技术,由内源性E3、透膜性好dTAG 合成小分子、外源变异非天然蛋白FKBP12F36V与需降解的目标蛋白融合体三部分组成。双功能的dTAG‑13等dTAG化合物可高效选择性地将FKBP12F36V融合的靶蛋白募集到E3 CRBN 附近,形成一个三元复合体,并引导靶蛋白K48多聚泛素化和蛋白酶体的降解(图2d)。体外细胞实验证实,dTAG 降解技术可快速选择性降解与FKBP12F36V融合嵌合体,如ENL、MELK、BRD4、EZH2、HDAC1、KRAS、MYC、PLK1、SLC蛋白等[44]。体内实验中,该技术被成功用来降解小鼠模型IE2蛋白亚型,阐明其在人巨细胞病毒(HCMV)晚期感染中作用[45];Nabet等[46]2020 年开发第二代dTAGv‑1 分子,可选择性与VHL E3 复合体结合(图2d),解决了影响一代dTAG‑13诱导靶蛋白有效性的背景特异性和蛋白特异性问题。小鼠体内药代动力学和药效学表征发现,与dTAG‑13相比,dTAGv‑1半衰期更长、暴露量更大、蛋白降解持续时间也更长[46]。
1.5 AID(auxin-inducible degron system)
植物生长激素(auxin)可诱导细胞通过蛋白酶体降解抑制生长素/吲哚乙酸(AUX/IAA)蛋白家族的转录抑制因子,激活编码AUX/IAA 家族蛋白基因表达,促进植物生长发育。AID系统的开发正是基于将植物生长素依赖性降解途径移植至其他真核物种中,由E3 SCF 复合体、靶蛋白和植物激素三个基本单位组成。TIR1 是一种特殊F‑box 蛋白,可选择性识别AID 标签序列(来源于AUX/IAA)标记内源靶蛋白(图2e)。该系统已成功应用于包括酵母、果蝇、秀丽隐杆线虫及源于鸡、小鼠、仓鼠、猴子和人类的细胞系中[47‑48]。当IAA生长素被供应时,TIR1 蛋白的激活可诱导靶蛋白快速耗竭,半衰期为10~20 min,大多数靶蛋白3 h可降解至不可检测水平[49],并可通过去除生长素来逆转[47]。近年来,AID 技术也进行不断改进,比如用合成生长素1‑萘乙酸(NAA)替代吲哚乙酸(IAA)避免GFP激发时IAA的光破坏形成有毒衍生物和生长缺陷[50];建立基于CRISPR‑Cas9 基因编辑技术的AID标签与靶基因融合表达技术,并开发最小化7 ku AID 标签(m‑AID),方便融合蛋白形成[51];开发双顺反子一体化质粒,可同时表达TIR1 和AID 标签融合靶蛋白,减少基因操作[52]。
1.6 Trim-Away
Trim‑Away技术是2017年Melina Schuh团队基于Leo James发现的Trim21蛋白而发明的一种在细胞内快速降解特定蛋白的新技术[53]。E3 Trim21与抗体Fc 区域具有很强亲和力,且在多种细胞类型和组织中广泛表达,可引入外源Trim21 用作抗体受体与针对靶蛋白的抗体Fc 段结合,招募UPS 体系来降解与抗体结合的靶蛋白(图2f)。Schuh 等证实应用Trim‑Away技术可以降解哺乳动物细胞和小鼠与人原代细胞中不同目标靶蛋白,及降解定位于不同细胞区域靶蛋白。并且通过设计特异性抗体,也可用于选择性降解翻译后修饰蛋白、剪切或突变的蛋白质变体,而保留未修饰的/野生型蛋白。比如,在NIH 3T3 细胞内成功将绿色荧光蛋白降解,半衰期仅为16 min。对细胞质中定位于质膜或囊泡样结构的膜锚定GFP、定位于染色质的组蛋白H2B‑GFP、积累于细胞核内的核定位NLS‑GFP 等均实现快速降解,半衰期分别为21 min、9 min、9 min。在NIH 3T3 和卵母细胞中,HTT 蛋白的致病突变体(3B5H10)被迅速降解,而正常的野生型HTT 蛋白不受影响。唐开福教授等[54]证实Trim‑Away技术可用来研究斑马鱼胚胎发育初期几小时的蛋白质功能,相比斑马鱼最为常见的基因敲降技术反义吗啉环寡核苷酸(morpholino)需数小时甚至数天,Trim‑Away技术仅10 min即可剔除目标蛋白,因此可用来研究母源性蛋白在胚胎发育早期(比如受精后2~3 h)的功能。
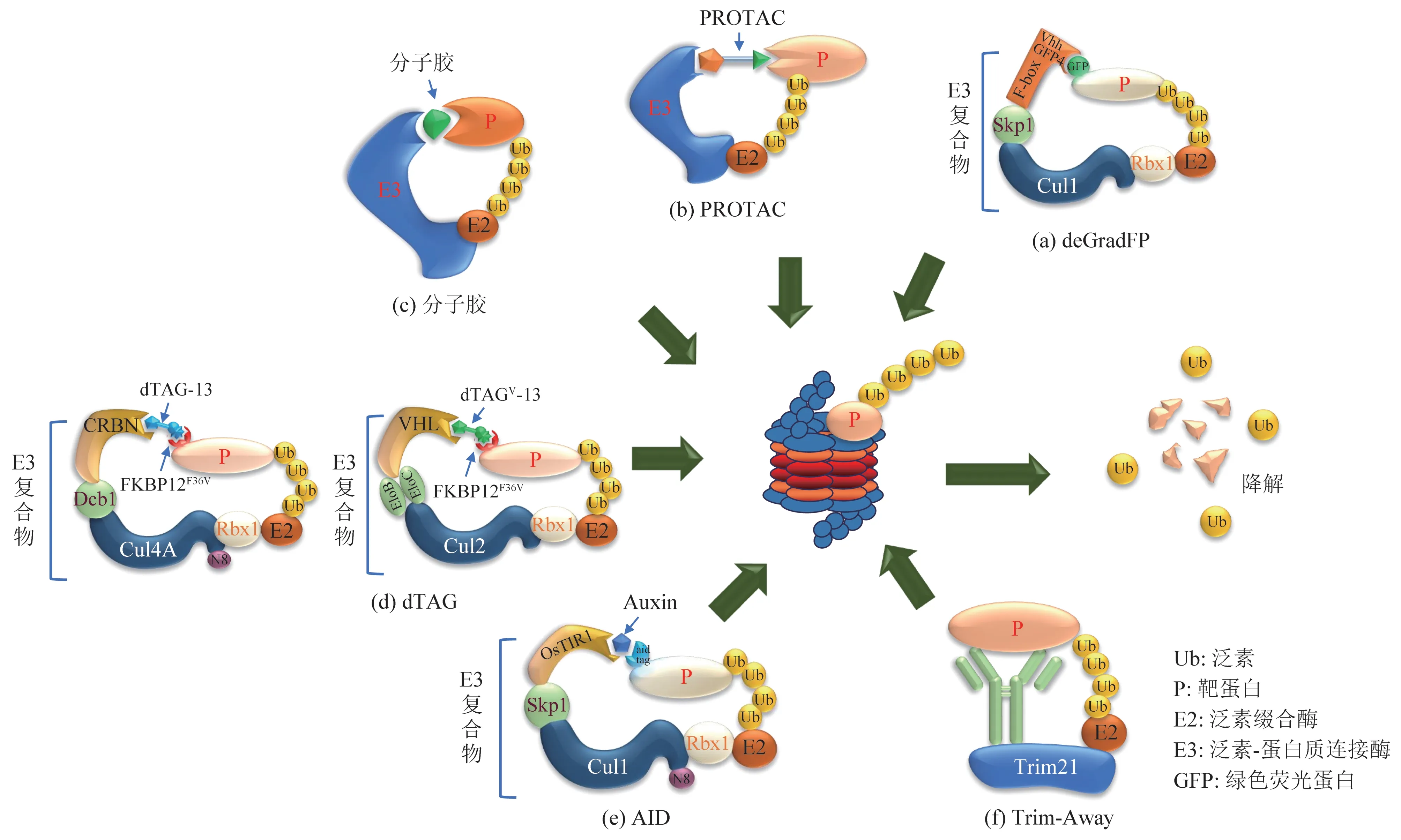
Fig.2 Protein degradation technology based on UPS图2 基于UPS蛋白质降解技术
2 基于自噬途径(autophagy)蛋白质降解技术
细胞自噬是细胞内部将受生理和病理因素影响而受损、变性、老化或失去功能的细胞、细胞器、蛋白质或核酸等生物大分子进行分解代谢,是广泛存在于真核细胞内的一种自我保护机制。细胞受自噬诱导信号后,在相关基因调控下,在被降解物附近形成小的脂质体样双膜结构,脂质化的LC3 蛋白附着脂质双层膜上,形成吞噬载体;吞噬载体膨胀吞噬带降解蛋白质或细胞器形成囊泡状的自噬体(autophagosome),随后与溶酶体(lysosome)融合形成自噬溶酶体(autolysosome),由溶酶体内水解酶降解包裹内容物,生成的氨基酸、脂肪酸等被输送到胞浆中,供细胞重新利用,以实现细胞代谢和能量的更新,维持细胞稳态。基于自噬途径,科研工作者开发出自噬靶向嵌合体(autophagy targeting chimera,AUTAC)、自噬栓系复合物(autophagosome tethering compound,ATTEC)蛋白降解技术。
2.1 AUTAC
真核细胞在受链球菌入侵后,链球菌蛋白会被K63多聚泛素化引起自噬降解。研究表明,S‑鸟苷酸化修饰是K63多聚泛素化修饰的前置步骤,推测S‑鸟苷酸化修饰很有可能是一种特异性的自噬信号。基于此,2019年日本Hirokazu Arimoto研究团队[55]开发AUTAC 技术。AUTAC 的设计与PROTAC相似,采用的是“两端靶向小分子+中间Linker”的模式,一端为特异性识别靶蛋白的配体,一端为模仿S‑鸟苷酸修饰的降解标签(即鸟嘌呤衍生物)。AUTAC也通过E3泛素化发挥作用,与引起蛋白酶体降解的K48多聚泛素化不同,其泛素化修饰是被选择性的自噬途径识别的K63多聚泛素化(图3a)。该研究团队已设计出系列靶向MetAP2、FKBP12、BET、TSPO 等蛋白质的AUTAC 分子,并成功进行降解。同时,还通过靶向定位于线粒体表面的TSPO 蛋白(mitochondrial translocator protein),成功引起线粒体膜蛋白TSPO的K63多聚泛素化修饰和破损线粒体降解。
2.2 ATTEC
ATTEC 技术是复旦大学鲁伯埙教授于2019 年发明[56]。与PROTAC 和AUTAC 不同,ATTEC 分子不依赖泛素化,不需要Linker介入,通过直接结合靶向蛋白和关键的脂质化自噬小体蛋白LC3(图3b),将靶蛋白与自噬小体捆绑在一起,从而引起自噬降解。鲁伯埙教授团队从3 375个小分子化合物库中成功筛选到2 个小分子10O5、8F20,可同时结合LC3 及突变HTT 蛋白,但不与野生型HTT蛋白结合,同时进行构型合理优化,又成功合成2个新小分子化合物AN1 和AN2。在细胞水平和亨廷顿病(HD)果蝇、小鼠动物模型上已成功验证了ATTEC的降解效果和疗效。该团队2021年新升级LD‑ATTEC 还具有降解包括自噬识别的非蛋白质生物大分子,如DNA/RNA分子、受损的细胞器等不同类型的靶标的巨大潜力[57],但ATTEC能否影响全身自噬还有待进一步阐明,以避免功能蛋白和细胞器的非特异性降解。

Fig.3 Protein degradation technology based on autophagy pathway图3 基于自噬途径蛋白质降解技术
3 基于内体-溶酶体途径(endosomelysome pathway)蛋白质降解技术
UPS系统与自噬途径的作用靶点均集中于胞内蛋白,对分泌蛋白和细胞膜蛋白无效。通常,胞外蛋白及细胞膜蛋白的降解由细胞表面溶酶体靶向受体(LTRs)家族通过内体‑溶酶体途径来促进蛋白质向溶酶体的转运。在溶酶体内的酸性环境下,60余种酸性水解酶如酶蛋白酶、核酸酶、糖苷酶、脂肪酶和磷酸酶等共同作用将外源蛋白降解成肽段。
斯坦福大学Carolyn R.Bertozzi教授团队[58]于2020 年开发设计出LYTAC(lysosome‑targeting chimaeras,溶酶体靶向嵌合体)技术,溶酶体靶向嵌合体LYTACs具有两个结合域:寡聚糖肽基团(oligoglycopeptide group)和可与靶蛋白结合的抗体或小分子,两者通过Linker连接。抗体或小分子可与胞外或者跨膜的目标靶蛋白结合,寡聚糖肽基团被细胞表面跨膜阳离子‑非依赖甘露糖‑6‑磷酸受体(CI‑M6PR,又称IGF2R)识别结合后,形成CI‑M6PR‑LYTAC‑靶蛋白复合物,可被细胞膜内吞形成运输囊泡转运至溶酶体,在溶酶体低pH 下,CI‑M6PR 自身由高尔基体再次转运到细胞膜循环利用,而靶向蛋白则被溶酶体内多种蛋白酶降解(图4)。利用LYTAC 技术已成功实现膜蛋白如表皮生长因子受体(EGFR)、程序性死亡配体1(PD‑L1)、转铁蛋白受体(CD71)以及分泌蛋白载脂蛋白E4(APOE4)的降解[58]。
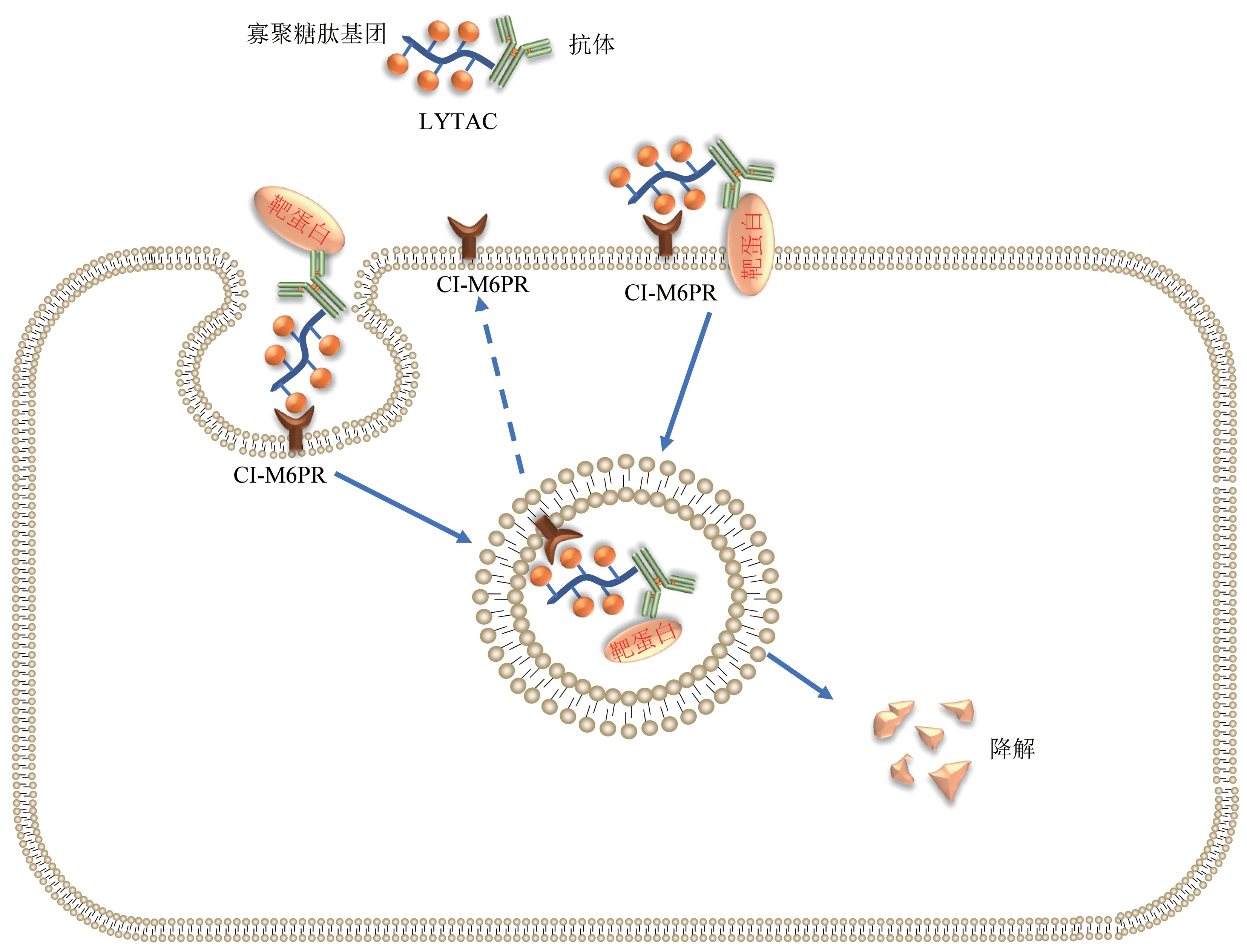
Fig.4 Protein degradation mechanism of LYTAC图4 LYTAC 蛋白质降解机制
4 靶向蛋白质技术应用与药物开发
继2009 年Nurix Therapeutics 成立后,Arvinas(2013 年,PROTAC 发明人成立)、C4 Therapeutics(2015年)、Kymera Therapeutics(2015年)等公司相继成立,致力于探索小分子蛋白降解剂开发和治疗研究。不同靶向蛋白质降解技术均具有自己的优势和劣势(表1),其中PROTACs研究的最为深入和详细,研究领域主要集中在肿瘤疾病,但在神经退行性疾病、炎症和免疫学领域也有所突破。
2019年,首个靶向AR的PROTAC口服小分子ARV‑110进入临床试验,标志着PROTAC技术迈出了成药性关键一步,成为该领域的里程碑事件。目前,已有9 种PROTACs 正进行临床试验[59](表2),主要针对肿瘤适应证。根据公司官网管线介绍,Kymera 公司KT‑413 和KT‑333、Nurix 公司NX‑5948 将于2021 年进入I期临床,Cullgen 公司CG001419、C4 Therapeutics 公司CFT8634 将于2021 年提交新药临床试验申请[59]。我国四川海思科制药有限公司研发的I类创新化学药——口服PROTAC小分子抗肿瘤药物HSK29116药品临床试验申请获得国家药品监督管理局受理(临床登记号CTR20210774)。HSK29116 能诱导BTK 泛素化标记,通过蛋白酶体途径将其降解,从而阻断BCR 信号通路的传递,抑制B 细胞淋巴瘤细胞的生长与增殖,起到双重抗肿瘤作用。苏州开拓药业股份有限公司外用雄激素受体(AR)降解剂GT20029(凝胶/酊)于2021 年4 月正式获得国家药品监督管理局颁发的临床试验通知书,2021年7月获美国食品药品监督管理局的临床试验许可,成为全球首个进入临床阶段的外用PROTAC 药物,用于雄激素性脱发和痤疮的治疗。

Table 1 Comparison of different protein degradation technologies表1 不同蛋白质降解技术对比
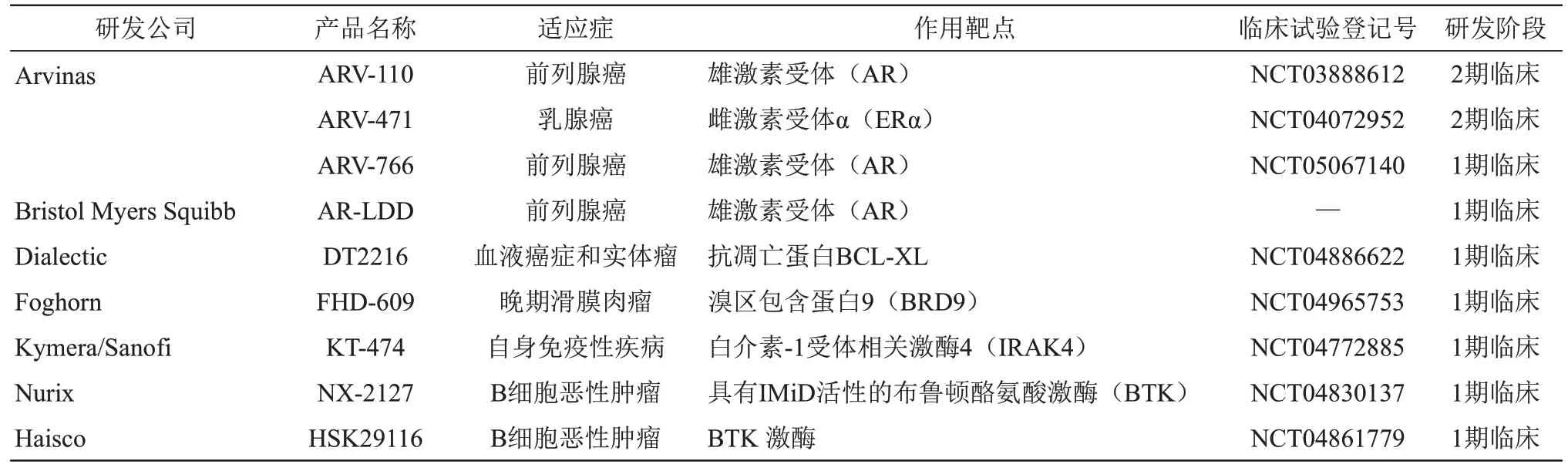
Table 2 Part of PROTAC has entered the clinic表2 部分已进入临床的PROTAC
虽然FDA 已批准ARV‑110 等PROTACs 进入临床研究,但目前仍缺乏PROTAC 在动物层面的临床前研究,尤其是大动物上的评价。清华大学饶燏课题组等[60]首次通过化学设计PROTACs,构建系统性敲除模型,成功实现小鼠、大鼠、猪和恒河猴全身体内FKBP12蛋白的快速可逆的敲低,其中小鼠及大鼠体内FKBP12 蛋白仅用1 d,恒河猴体内FKBP12 蛋白仅用3 d 即可高效率全身敲低(甚或敲除),首次验证PROTACs可采用口服给药途径保持高效蛋白质降解功能,且发现停止给予PROTACs,动物体内FKBP12蛋白可逐渐恢复,这有利于建立动物模型自身对照,更为准确研究蛋白质功能。将此方法应用于BTK 蛋白全身敲低,也获得显著效果,充分证实该技术可行性。该工作实现了PROTAC 技术从小鼠、猪到恒河猴的系统探索,填补了PROTAC 技术临床前研究的空白,为其应用于临床治疗人类疾病提供了指导,奠定了坚实基础。饶燏课题组[61]于2020年又进一步首次在小鼠体内揭示PROTAC 对FAK 蛋白非激酶结构域的调控,并系统评价了FAK 靶向的PROTAC 分子在动物水平的生物学功能,研究表明,PROTACs可用于开展动物体内蛋白质非酶活功能的研究,弥补了传统小分子抑制剂只作用于蛋白酶活功能的缺陷。
5 结论与展望
靶向蛋白质降解技术具有无需结合到靶蛋白的活性位点/口袋、高靶点选择性、低暴露量即可发挥有效作用等优势,在蛋白质靶点确认及填补靶向“不可成药”蛋白的空白方面,展示出极大应用前景。但新技术的出现,尚存在诸多不足与挑战,例如,人体内存在有600 余种E3,然而可用于PROTAC设计的E3尚不到10种,拓展可用靶向蛋白质降解的E3或开发不依赖E3的降解方式是面临的严重挑战之一;靶向蛋白质降解技术的特异性和潜在的非目标效应亟需确证;靶向蛋白质降解也存在耐药性问题,实验研究发现肿瘤细胞可通过降低E3的活性及提高E3其他底物的水平等多个途径产生对蛋白质降解剂产生耐药;成药性评价体系缺乏也急需面对,比如,PROTAC可循环催化,实现彻底地降解靶向蛋白质,传统方法无法准确评估PROTAC的药代动力学和药效动力学性质,以及是否存在潜在的严重毒性。
靶向蛋白质降解技术现已实现靶向降解胞外、细胞膜上和胞内等不同定位的游离蛋白质。基于溶酶体LYTAC、AUTAC 和ATTEC 降解途径甚至可靶向降解一些蛋白质聚集物、编码致病蛋白的遗传物质以及清除致病细菌和病毒等。随着蛋白质降解技术的不断发展与完善,如Snipers[62‑63]、ENDTACs[64]等新技术出现,极大扩展靶向蛋白质降解技术的目标范围和潜在应用,加上多项临床试验积极结果也证实靶向蛋白质降解药物巨大开发潜能,靶向蛋白质降解技术已被视为未来人类新药开发最重要的方向之一。持续优化现有降解技术,探索更安全、高效、精准、可控的新型靶向蛋白质降解技术,为药物设计和疾病的靶向治疗提供新的思路,成为当前研究的主要方向。
