突变和修饰β淀粉样蛋白与阿尔茨海默病
2022-02-10郝秀萍武林芝
郝秀萍 武林芝
(太原学院材料与化学工程系,太原 030032)
阿尔茨海默病(Alzheimer’s disease,AD)是痴呆症的主要类型,是一种渐进性脑部疾病,它逐渐破坏人的记忆和思维能力,最终导致日常行为能力的丧失。在影响病人生活质量的同时,还带来源源不断的社会问题,是目前最受关注的神经和精神疾病之一[1]。AD 的两大病理特征包括由tau 蛋白形成的细胞内神经原纤维缠结和由β 淀粉样蛋白(Aβ)形成的细胞外老年斑[2‑3]。人们认为Aβ的错误积累在AD 的病理学中起着核心作用[4]。以Aβ为靶标的疗法主要针对的是单体或寡聚体的Aβ40和Aβ42,但以Aβ40 和Aβ42 为靶标的临床治疗结果并不理想,人们对此进行了分析并提出了多靶标策略[5]。淀粉样前体蛋白(amyloid precursor protein,APP)的水解加工过程会产生一系列不同长度的截短Aβ种类。此外,Aβ肽还会受到广泛的翻译后修饰。虽然大多数AD病例是散发性的,但已经确定了许多家族性阿尔茨海默病(familial AD)和相关的脑淀粉样血管病会导致AD 的早发和严重程度的增加,许多FAD的Aβ会发生单个氨基酸突变。大量的研究表明,这些突变Aβ、截短Aβ、被修饰的Aβ会加快聚集动力学,改变聚集体构象,并具有更高的细胞毒性。这些Aβ 种类可能破坏了脑内稳态并加速了AD神经变性。我们调查了大量的文献,对这些Aβ 种类的聚集特性及在AD发展过程中可能产生的影响进行分类总结,并评述了检测分析这些Aβ种类的方法。
1 Aβ变体
大多数AD 病例是散发的,但一小部分称为FAD 的AD 病例与早老素1、早老素2 或APP 的突变有关。根据这些FAD 首次发现的地域,分别命名为英国型(English,H6R)、日本鸟取型(Tottori,D7N)、佛兰德型(Flemish,A21G)、大阪型(Osaka,E22Δ)、荷兰型(Dutch,E22Q)、意大利型(Italian,E22K)、北极型(Arctic,E22G)、爱荷华型(Iowa,D23N)、皮尔蒙特型(Piedmont,L34V)等。位于淀粉样蛋白序列内部的APP 突变会导致多种疾病表型,包括早发性痴呆、脑淀粉样血管病、出血性中风等。这些氨基酸取代改变了Aβ 的产生量,增加了Aβ42 与Aβ40 的比率,改变了突变Aβ 变体的聚集潜力,有的还促进了有毒聚集体的形成。表1 列出了不同Aβ 突变体肽的聚集特性及其影响。
1.1 突变Aβ肽的影响及相应临床表现
A2T突变是一种保护性的突变,A2T突变能减少β 分泌酶1(BACE1)对APP的裂解。体外研究表明,它使淀粉样蛋白聚集物的形成减少约40%,可以预防AD 和老年人的认知能力下降[6],但Aβ 42A2T 肽的体外细胞毒性高于野生型Aβ42,并且在聚集的早期与模型膜相互作用[7]。A2V 变异在纯合子个体中是致病的,杂合子的亲属没有受到影响,隐性A2V 突变能增加Aβ 的产生[8],A2V Aβ的沉积物很大,主要存在血管周围,在影响小脑的同时会减少新纹状体[9]。English(H6R)Aβ 的细胞毒性更强[10],H6R 突变导致His6 结构域不能参与锌离子的鳌合,而由(EVHH14)‑E‑11 片段配位的锌离子能促进其形成稳定的二聚体[11]。
人们在一个可能患AD的日本家系中,发现了D7N错义突变[12]。Aβ D7N突变增强了寡聚体的细胞毒性[10]。Chen等[13]在一个具有早发性AD的台湾家庭中,鉴别出了位于Aβ 序列内的新型APP 突变——D7H,这种突变增加了Aβ 的产生,提高了Aβ42/Aβ40 的比率,具有更高的神经毒性。由于D7H 突变型Aβ 具有一个额外的金属离子配位残基组氨酸,它与Zn2+/Cu2+具有更高的亲和力,这可能促使Aβ D7H 具有高致病性。BACE1 酶是Aβ 产生过程中一种非常重要的切割酶,它有两个切割位点,β 位点(1 号位点)和β′位点(11 号位点),E11K 突变使得BACE1 切割位点移向了β 位,导致Aβ40 和Aβ42 的水平含量增加了2~3 倍,Aβ42/Aβ 40的比率也略微升高。
APP 687 位的赖氨酸‑天冬酰胺取代(根据Aβ编号称为K16N),导致早期具有常染色体显性遗传模式的痴呆。K16N突变位于α分泌酶切割位点,使得APP 的分泌受到影响,产生了更多的Aβ 肽。带有K16N 突变的Aβ 肽的独特之处在于该肽本身对神经元细胞无害,然而野生型和突变体肽的等摩尔混合物则显示出很强的毒性。此外,Aβ42 K16N抑制野生型Aβ42 原纤维的形成。Aβ42 K16N 不易被主要的Aβ降解酶脑啡肽酶清除[14]。
Flemish(AβA21G)AD 是在一个具有早老性痴呆和脑淀粉样血管病引起的脑出血家族中发现的遗传突变类型[15]。与其他以脑Aβ42为主的FAD病例相反,Flemish AD 病人主要沉积Aβ40,而且大脑中的老年斑主要集中在血管上。Yagi‑Utsumi 和Dobson[16]的研究表明,A21G突变改变了Aβ40形成淀粉样蛋白原纤维的初始成核过程,从而影响了Aβ 的膜结合特性。Aβ A21G 肽被脑啡肽酶降解的速度明显比野生型Aβ 或任何其他突变体肽慢[17]。Aβ42 A21G 和Aβ42 野生型都能诱导神经元细胞凋亡,特别是N 端截短肽毒性更强[18],但Wang等[19]对Aβ42 A21G的体外细胞实验表明,从细胞形态、生存力或细胞增殖来判断,Aβ A21G 和Aβ野生型对两种类型的平滑肌细胞均未引起明显的毒性。
在一个患有痴呆症及AD的日本家系中确定了Aβ E22∆,即缺少22 号谷氨酸的变体Aβ[20]。该突变显著降低了总Aβ的分泌,但变体Aβ对蛋白质水解的抵抗力更高,并且比野生型Aβ 更有效地抑制大鼠海马长时程增强。与无毒Aβ40 野生型相比,Aβ40 E22Δ在大鼠原代神经元培养物中具有神经毒性,而Aβ42 E22Δ 的毒性低于Aβ42 野生型,但在神经元原代培养物中,每个细胞的神经突生长明显减少[21]。E22G 是在患有AD 的瑞典家庭中发现的突变类型,这种突变的携带者显示血浆中Aβ42 和Aβ40水平降低[22],聚集过程早期形成的中间物会从神经突开始引发细胞功能障碍,从而导致神经元损伤,并引起中度的神经胶质和炎症组织反应[23‑24]。Aβ E22G 突变使其对脑啡肽酶具有抵抗力,因此,Aβ E22G 不仅可以通过促进原纤维形成,而且可以通过延长Aβ 在大脑中的半衰期来致病[25]。Bugiani 等[26]对患有淀粉样变性与Aβ E22K 突变相关的遗传性脑出血患者的临床研究显示,受影响的个体表现为反复发作的头痛和多发性中风,大多数患者随后出现癫痫和认知能力下降。Aβ E22K 突变使其对脑啡肽酶也具有抵抗力[25]。在患有淀粉样变性的遗传性脑出血的荷兰患者,即淀粉样变性‑荷兰型(HCHWA‑D)中,Aβ 22 位的氨基酸发生谷氨酰胺(Q)取代谷氨酸(E),导致了Aβ 在这些患者的脑血管壁中沉积,从而导致脑出血和过早死亡[27]。Aβ E22Q 对脑啡肽酶也具有抵抗力[25],Aβ40 E22Q 会在培养的人脑血管平滑肌细胞中诱导强大的病理反应,包括提高APP 水平和细胞死亡[28]。在APP Dutch小鼠和HCHWA‑D人脑中,Aβ40与Aβ42的比率显著高于APP野生型小鼠或AD 人脑中的比率[29]。野生型Aβ 容易从脑中转运到血浆中,但荷兰型突变体Aβ 不容易清除出去,这可能导致其在脑血管系统中的大量积累[30]。另外,Aβ E22Q 刺激基质金属蛋白酶2(MMP‑2)的表达和激活,而MMP‑2 的表达和激活增加可能会导致人脑血管平滑肌(HCSM)细胞因致病性Aβ而死亡[31]。
Grabowski 等[32]报告了爱荷华州三代家庭中APP 的一个新突变位点,694 位(对应于Aβ 的23号)天冬氨酸被天冬酰胺取代(D23N),是常染色体显性痴呆。患者伴有进行性失语性痴呆,白质脑病和枕骨钙化,神经病理学检查发现严重的脑淀粉样血管病,广泛的神经原纤维缠结和斑块中Aβ40的异常分布。研究表明,D23N突变显著增加了神经元和内皮细胞的肽毒性[33]。
Obici等[34]报道了一个常染色体显性遗传、复发性脑出血的家庭中Aβ 序列内的新型突变(L34V),病理检查发现该突变导致了严重的脑淀粉样血管病,但没有实质性淀粉样斑块或神经原纤维缠结。L34V Aβ形成的寡聚体或原纤维能诱导人脑微血管内皮细胞和平滑肌细胞凋亡[35]。
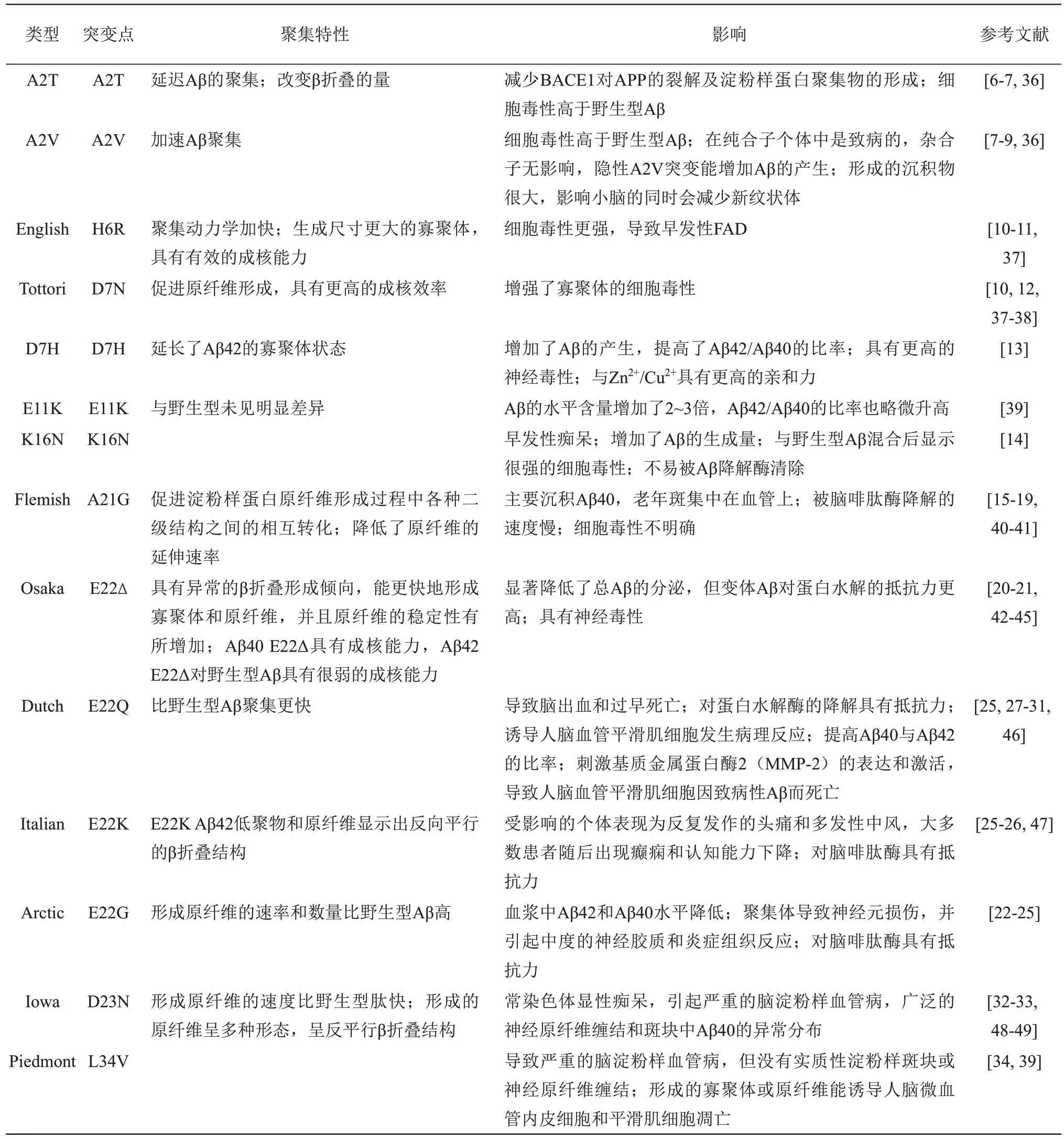
Table 1 A compilation of aggregation characteristics and effects of amyloid β mutant peptides表1 β淀粉样蛋白突变体肽的聚集特性及其影响
1.2 突变Aβ肽的聚集特性
A2T 突变是一种保护性的突变,能够延迟Aβ肽的聚集[36],显著改变β 发夹的量并打破其转变为其他结构的平衡[7],而A2V突变会加速Aβ肽的聚集,使其更快地演变为β 折叠构象[7,36]。H6R突变能在淀粉样原纤维形成的延伸阶段促进原纤维的形成[37],H6R突变Aβ加快了聚集动力学,使得Aβ 的二级结构更快地由无规则卷曲结构转变为β折叠结构,并生成尺寸更大的寡聚体,这些寡聚体具有有效的成核能力。D7N 突变体能在淀粉样原纤维形成的延伸阶段促进原纤维的形成[37],并且具有更高的成核效率[38]。E11K Aβ与野生型Aβ的聚集动力学、聚集体形态及细胞毒性未见明显差异[39]。
A21G 突变可以增加Aβ 的分泌,并改变其结构[40],促进富含β 折叠的淀粉样蛋白原纤维形成过程中各种二级结构之间的相互转化[41]。Betts等[17]的研究表明,A21G 突变降低了原纤维的延伸速率。E22∆Aβ表现出增强的寡聚特性但没有原纤维化的聚集特性[20],Inayathullah 和Teplow[42]对E22Δ Aβ40和E22Δ Aβ42的研究结果阐明了这些突变体肽的构象动力学、原纤维形成动力学、原纤维形态和原纤维稳定性,表明两种E22Δ Aβ肽均具有异常的β折叠形成倾向,能更快地形成寡聚体和原纤维,并且原纤维的稳定性有所增加。此外,E22Δ Aβ42 低聚物似乎与野生型Aβ42 低聚物的大小、形状均不同[43]。E22Δ Aβ40 具有成核能力,诱导野生型Aβ40 形成原纤维,这些原纤维比同源接种的Aβ40 原纤维的稳定性差,但是,E22Δ Aβ 42 对野生型Aβ 具有很弱的成核能力[44‑45]。E22G Aβ 形成原纤维的速率和数量都比野生型Aβ 高得多[22]。体外聚集实验表明,野生型和E22K Aβ 均形成了以交叉β结构为特征的原纤维,但具有明显不同的β折叠结构,与野生型原纤维的平行β折叠结构相比,E22K Aβ42低聚物和原纤维均显示出反向平行的β 折叠结构[47]。分子动力学模拟研究表明,E22Q Aβ 比野生型Aβ 聚集更快[46]。D23N Aβ 40 形成原纤维的速度比野生型快得多,电子显微镜显示,D23N Aβ40 形成具有多种形态的原纤维,并且大多数原纤维呈反平行β折叠结构[48‑49]。
2 Aβ修饰肽
根据淀粉样蛋白级联假说,Aβ 的聚集在AD的发展过程中起关键作用。在FAD患者中,Aβ 的聚集是由遗传突变引起的,但是大约90%的AD患者是散发性的,什么原因导致了散发性AD患者中Aβ 的聚集,目前还不明确。对老年斑组成的分析表明,聚集的Aβ 以不同的方式被修饰,主要是通过Aβ 的截短和后修饰,其中,截短肽包括从氨基酸残基A2 和焦谷氨酰化E3、F4、R5、H6、焦谷氨酰化E11 开始的N 端截短肽,以及长度为16、24、34、37~43的C端截短肽。我们之前的研究总结了Aβ 截短肽的生理和病理特性[50]。Aβ 的修饰主要包括焦谷氨酸化、天冬氨酸异构化、氧化、磷酸化、硝化、瓜氨酸化等。过多的修饰表现出致病特征,如聚集增加、神经毒性增加、抑制海马体长时程增强的能力等。表2 列出了不同Aβ 修饰肽的聚集特性及其影响。
2.1 Aβ修饰肽的产生与鉴定
从3 号或11 号谷氨酸开头的N 端截短Aβ 肽容易发生焦谷氨酸化。从AD患者的神经斑和脑血管壁中,纯化并定量了以焦谷氨酰基(Aβ pE3)形式从E3 残基开始的Aβ 肽,N 端截短的Aβ pE3 占神经斑块中Aβ 的51%[51‑52]。Sullivan 等[53]使用特异性抗体在老年痴呆症大脑皮层的老年斑中识别到了Aβ pE3和Aβ pE11,并且Aβ pE11是最内层核心的主要形式。蛋白质或肽中天冬氨酸和天冬酰胺的异构化通过在生理条件下经由琥珀酰亚胺作为中间体的非酶促反应产生L‑异天冬氨酸。已经在体内多种胞内蛋白以及神经退行性脑组织中病理沉积的蛋白中鉴定出了异天冬氨酸[54‑55]。在7 位或23 位异构化的Aβ 肽差异性地沉积在AD 大脑的老年斑和血管淀粉样蛋白中。氧化应激和淀粉样斑块的形成是AD 的两个关键标志。Aβ 中35 位的一部分甲硫氨酸(Met)在AD 大脑斑块中被氧化成甲硫氨酸亚砜(Met(OX))。在AD 大脑中发现了磷酸化淀粉样蛋白,使用反义肽方法,确定了人类细胞周期蛋白依赖性激酶1(CDK‑1)负责Aβ 的磷酸化[56]。细胞外的Aβ在细胞表面和人脑的脑脊液中被蛋白激酶磷酸化。Kumar 等[57]开发了新的抗体来根据S26 的磷酸化状态特异性区分Aβ 肽,利用新抗体,在神经元内和脑血管中发现大量的磷酸化Aβ。AD 的部分炎症反应是诱导型一氧化氮合酶NOS 的上调,导致NO 产生增加,NO 通过诱导翻译后蛋白质修饰促进细胞信号传导。在病理条件下,信号转导作用转变为过氧亚硝酸盐和二氧化氮等副产物形成蛋白质酪氨酸硝化作用。Aβ 是NO靶标之一,其在Y10 处被硝化[58]。瓜氨酸化和脱酰胺是衰老相关的蛋白质翻译后修饰,在中性pH值下会增加Aβ 上的负电荷数量[59]。在AD 大脑所有已识别的蛋白质翻译后修饰类型中,乙酰化仅影响总修饰肽的约10%,但乙酰化的Aβ和tau聚集体水平增加最高[60]。Aβ 肽有两个潜在的乙酰化位点,K16和K28。
2.2 Aβ修饰肽的聚集特性
含焦谷氨酰的肽比相应的全长Aβ 肽更容易形成β折叠结构,并且更稳定,聚集倾向更高,诱导的细胞毒性更强[61‑62]。体外实验表明,23 位的异构化修饰大大增强了Aβ的聚集。23位的自发异构化诱导Aβ发生构象变化形成β转角,β转角结构在Aβ 肽的聚集和神经毒性中起着至关重要的作用[63‑64]。Met35侧链氧化成Met35(OX)显著阻碍了生理pH下Aβ42的原纤维形成的速率,减少了两种主要形式的Aβ 产生的纤维总量,Met35(OX)还可以改变Aβ原纤维的形态并阻止原纤维的形成。甲硫氨酸氧化的Aβ40和Aβ42的纤维形态发生了明显的变化,纤维长度明显减少[65]。也有研究显示Met(OX)的存在减少了Aβ40 纤维形成的滞后时间,但延长了Aβ42 的滞后时间[66]。8 号丝氨酸残基的磷酸化促进了寡聚Aβ 组装体的形成[67]。Jamasbi 等[68]研究了磷酸化Aβ42 合成肽的二级结构、聚集特性以及与初级皮层神经元质膜的相互作用,结果表明在合成脂质环境中,磷酸化Aβ42 增加了β 折叠形成,能快速形成聚集体。Aβ 的硝化加速了其聚集,并在APP/PS1 小鼠和AD 大脑的Aβ 斑块核心中检测到硝化Aβ[58]。也有研究表明,酪氨酸硝化显著降低了Aβ40的聚集[69]。这种积极作用可能是由于在生理pH 值下硝化引起带负电荷的Y10 与E11 和H6 或H13 之间的盐桥相互作用并阻断Y10 周围的扭结,从而防止Aβ 纤维化和聚集[70]。Osaki等[59]研究了瓜氨酸化和脱酰胺对Aβ纤维化特性的影响,结果表明Aβ40 的R5→Cit 修饰不影响原纤维化率,并形成与Aβ40 原纤维不同的β折叠结构。Aβ40的N27→D修饰阻碍了原纤维化并诱导了反平行β 折叠聚集体的形成。具有R5→Cit修饰的Aβ42在水性介质中的溶解度增加,其原纤维形成速度比Aβ42 慢,但没有改变原纤维的平行β折叠结构。带有N27→D修饰的Aβ42部分形成了包含平行β折叠结构的原纤维。K28的乙酰化(K28Ac)减慢了Aβ42原纤维化速率但不影响原纤维形态。另一方面,K16 残基的乙酰化(K16Ac)大大降低了Aβ42 肽的原纤维化特性,也影响了其毒性[71]。
2.3 Aβ修饰肽的影响
Aβ pE3 的存在具有重要的结构意义,因为它比其他形式的Aβ更具疏水性,因此增加了Aβ的不溶性。此外,Aβ pE3 的N 端受普通氨基肽酶作用的阻断,可能导致Aβ 在AD 的神经斑块中大量积累。Aβ pE3的细胞毒性更强,AβpE3‑42对Aβ42原纤维的形成具有抑制作用,与其已知的更大感染力相一致[72]。Youssef 等[73]进行的小鼠实验表明,Aβ pE3‑42 可以介导在临床前AD 阶段发生的神经退行性过程和随后的认知改变。Met35的氧化态会影响先前存在的淀粉样蛋白原纤维和噬斑的稳定性,导致细丝、原纤维和成熟原纤维的形态发生变化[74]。与未氧化肽的神经毒性相反,氧化的Aβ在皮质神经元培养物中完全缺乏神经毒性作用[75],这可能是由于氧化态的Aβ 增强了甲硫氨酸亚砜还原酶A 型(MsrA)基因的表达和功能来减少活性氧ROS 的生成[76]。磷酸化的Aβ 肽显示出增加的神经毒性和降低的形成刚果红阳性原纤维的能力[55]。在转基因小鼠和AD 患者大脑中检测到磷酸化Aβ,在果蝇模型中,磷酸化Aβ显示出更高的毒性[67]。Aβ 的磷酸化可以在Aβ 代谢中发挥双重作用,一方面它降低了其蛋白水解清除率,另一方面促进了其聚集[77]。细胞毒性实验证实野生型Aβ 40比Aβ40Y10(3N)T更具细胞毒性[69]。对Aβ42的研究也表明Y10 的硝化可能会阻止Aβ42 的π‑π堆积相互作用,从而抑制其聚集和神经毒性[78]。Zhao 等[79]还研究了Aβ42 硝化与有毒Cu(II)的相互作用,结果表明尽管硝化没有改变Aβ42与Cu(II)的结合或Aβ42‑Cu(II)复合物的过氧化活性,但硝化改善了Cu(II)诱导的Aβ42 聚集和神经毒性。与单独的野生型或K28Ac肽聚集体相比,K16Ac 和双乙酰化(KKAc)肽聚集体显示出更高的细胞毒性。然而,野生型和乙酰化Aβ42 肽聚集体的异质混合物表现出更高的自由基形成和细胞毒性[71]。
3 Aβ变体的检测
近年来,关于Aβ 的分析检测方法研究取得了诸多进展,研究者从不同的角度对其进行了评述[80‑82]。由于Aβ40 和Aβ42 是人体内Aβ 的主要存在形式,目前建立发展的检测分析方法主要针对单体状态或聚集状态下的Aβ40 和Aβ42,关于Aβ 变体的检测方法报道的比较少。随着人们对AD病理特征的深入研究,发现了越来越多的Aβ 变体起着重要作用,可作为AD 的生物标志物。针对这些Aβ 变体,开发出了一些分析检测方法,包括常见的质谱法、免疫学法和电化学法等。
3.1 质谱法(MS)
质谱法因其准确度和精密度而成为一种可靠的检测Aβ 方式。由于Aβ 的疏水性、高分子质量(>4 ku)和低丰度,使用传统MS 方法(尤其是在血浆中)对Aβ 进行定量仍然是一个挑战,因此,对Aβ 的质谱分析通常与其他分析处理方法如免疫沉淀(IP)法、固相萃取(SPE)法、毛细管等电聚焦(CIEF)法等结合使用。
Domingo 等[83]开发了一种基于固相萃取和电喷雾电离液相色谱质谱(ESI‑LC‑MS)的无抗体方法,用于在人脑脊液(hCSF)中同时鉴定和定量19 种Aβ 亚型(Aβ1‑42、1‑40、1‑38 和16 种Aβ N端截短和翻译后修饰形式(包括焦谷氨酸形式)的肽。Portelius 等[84]描述了一种方法,它采用免疫沉淀结合基质辅助激光解吸/电离飞行时间质谱来确定脑脊液中C 端截短的Aβ 肽的模式,使用与磁珠偶联的抗体,在脑脊液中检测到18个C端和2个N 端截短的Aβ 肽,其中3 个最强的峰对应于Aβ1‑40、Aβ1‑17 和Aβ1‑38。Murayama 等[85]生成了一种新的单克隆抗体,该抗体对Aβ5‑40/42 的N端具有特异性。蛋白质印迹证实该抗体识别Aβ5‑40 但不识别Aβ1‑40。用抗体进行免疫沉淀,然后进行质谱分析,进一步检测到来自表达半胱天冬酶切割的APP 的神经母细胞瘤细胞的条件培养基中的Aβ5‑40。Kaneko 等[86]通过串联质谱法(基质辅助激光解吸/电离飞行时间质谱)对人血浆中进行鉴定,检测到22 种Aβ 相关肽,开发了Aβ相关肽的定量分析,并证明了血浆稀释线性和定量所需的精确度。Haußmann 等[87]提出了一种通过毛细管等电聚焦免疫测定、分离和检测Aβ N 端截短肽的新方法。CIEF 免疫测定和免疫沉淀质谱分析确定,从残基R5 开始的肽是在细胞培养模型中存在BACE1 抑制剂的情况下产生的主要氨基末端Aβ变体。Pannee等[88]开发了一种基于免疫沉淀的方法,用于从人血浆中富集Aβ 肽。使用基质辅助激光解吸/电离飞行时间/飞行时间质谱法分析肽段以进行Aβ 分析和选择反应监测,进行Aβ1‑38、Aβ1‑40 和Aβ1‑42 的MS 定量。在Sargaeva[89]的研究中,电子捕获解离(ECD)傅里叶变换离子回旋共振质谱(FTICR MS)被应用于检测Aβ 肽中的天冬氨酸的异构化.
3.2 免疫学方法
基于抗原抗体特异性反应的免疫检测方法是定性、定量分析Aβ 肽的最常用、可靠和灵敏的方法之一。酶联免疫吸附分析(ELISA)、蛋白质印迹法、免疫组织化学法、免疫荧光标记、免疫染色法也常用来鉴别和检测Aβ变体。
针对不同的Aβ 变体,人们获得了不同的抗体(表3)。Caillava 等[90]获得了针对人Aβ34 C 端最后14 个氨基酸的兔多克隆抗体(称为α34)。蛋白质印迹实验表明,α34 血清的整个纯化IgG 片段仅识别Aβ34,而不识别其全长对应物Aβ40。ELISA证实了α34 对Aβ34 的特异性,并且具有高灵敏度(检测下限约为1µg/L)。Sullivan等[91]生成了两种针对Aβ pE物种的多克隆抗体,并使用它们来观察人淀粉样斑块的Aβ pE谱。通过蛋白质印迹分析和免疫组织化学对这些抗体的评估表明,它们在低浓度下(5µg/L)与靶标特异性结合。Albertini等[92]设计了一种免疫蛋白质组学检测方法,该检测在预活化表面的芯片阵列上使用单克隆抗体混合物(6E10+4G8),然后使用表面增强激光解吸电离‑飞行时间质谱(SELDI‑TOF MS)分析检测到15 个Aβ亚型。Acero等[93]生成并表征了一种抗Aβ pE3小鼠单克隆抗体(3B8),可识别AD患者脑组织和3xTg‑AD 转基因小鼠脑组织中的淀粉样蛋白聚集体。针对不同Aβ 片段的ELISA分析结果表明,无论焦谷氨酸修饰如何,都可以识别AβpE3 和Aβ3‑42中存在的两个构象表位。在AD转基因小鼠模型中,Aβ4‑x 先于Aβ pE(3‑x)在神经元内积累。新型Aβ4‑x免疫反应性抗体NT4X‑167[94]可以检测到来自截短Aβ 物种的高分子质量聚集体,并且与其他常见神经退行性疾病典型的聚集体没有交叉反应。Liu 等[95]使用与辣根过氧化物酶连接的抗体(P82,M11),利用ELISA在血管淀粉样沉积物中检测到未修饰的游离Aβ11‑40和焦谷氨酸修饰的Aβ11‑40。Mehta 等[96]生成和部分表征了针对AβpE3 的兔单克隆抗体(Rab mAb),发现生成的Rab mAb 对AβpE3 具有特异性,因为它与Aβ16、Aβ40、Aβ42、Aβ3‑11 和Aβ pE(11‑17)在ELISA中没有反应性。在蛋白质印迹中,AβpE3的最佳检测条件是抗体浓度为0.5 mg/L,该抗体的灵敏度足以在夹心ELISA 中检测10 ng/L 的AβpE3。Mohamadi 等[97]提出了一种用于检测Aβ 肽的集成微流控芯片,该设备利用免疫捕获和微芯片电泳分析不同截短的Aβ 肽(1‑37、1‑39、1‑40 和1‑42),并且能够检测低至25 ng的加在未稀释的脑脊液中的合成Aβ 肽。Klafki 等[98]对针对Aβ(‑3‑x)N 端的两种抗体进行了表征,开发了用于测量Aβ(‑3‑40)的夹心免疫测定方法,并通过毛细管等电聚焦免疫测定、蛋白质印迹分析和免疫组织化学评估了抗体选择性。两种单克隆抗体都检测到Aβ(‑3‑40),与Aβ1‑40或N端截短的Aβ变体没有明显的交叉反应。免疫测定对Aβ(‑3‑40)显示出高选择性,定量测定范围为22 ng/L~7.5µg/L。除了针对Aβ截短肽的抗体外,研究者还开发了针对Aβ 修饰肽的抗体,如针对磷酸化的S8(pS8 Aβ)的特异性抗体1E4E11[99],针对磷酸化的S26(pS26 Aβ)的单克隆抗体5H11C10[100],针对硝化的Y10(3NTyr10Aβ)的3NTyr10Aβ 抗血清[101],针对焦谷氨酸化的E3(pE3Aβ)的抗体337.48及针对异天冬氨酸化的IsoD‑Aβ的抗体22C8[102]。
3.3 电化学法
电化学方法通常采用生物传感器将生化反应转变为可定量的物理/化学信号,由于其高效性、成本低、高特异性和快速性,成为目前最广泛使用的技术之一。基于酪氨酸或丝氨酸的磷酸化和酪氨酸的硝化对肽分子电化学活性的影响,Suprun等[103]在碳丝网印刷电极上记录了带有未修饰残基、O‑磷酸化Y10 或S8、3‑硝化Y10 的Aβ16 的氧化和还原的方波伏安图,结果表明这些氨基酸残基的磷酸化和硝化都显著改变了Aβ16 肽的电化学行为,利用电化学方法可以直接检测Aβ 中的翻译后修饰。Lu 等[104]通过TiO2纳米刷(TiO2NB)基于溶液的水热生长来构建光电化学(PEC)传感器用于检测Aβ1‑28,表现出极大灵敏度,检测限为26.3µg/L,促进了一种简单、无标记、快速、灵敏和无创的方法,以克服用于AD 诊断的常规技术的局限性。表面修饰、蛋白质纳米结构作为新兴的生物纳米技术平台,为构建生物传感器提供了强大的工具[105],基于该平台的电化学检测方法将为Aβ 变体的检测提供更多的可能。
4 结论与展望
位于Aβ序列内的FAD突变似乎具有不同的生化特性和病理效应,并导致不同临床表现和发展。除了A2T 是一种保护性突变外,其他大多数突变会增加向寡聚体或原纤维的聚集率,更具神经毒性和病理性。FAD点突变会改变Aβ肽的聚集动力学,形成形态、构象上不同的淀粉样蛋白结构,这可能解释了与Aβ突变相关的疾病表型的异质性。
除了FAD 中存在的Aβ 突变体外,散发性AD中还有大量的其他Aβ 种类,有些是在APP 加工过程中产生的,有些是经过翻译后修饰产生的,或者是在某些细胞或细胞外区室中发现或产生的。Aβ的特定区域显然对其生理特性有不同的贡献,如Aβ的N端区域及某些氨基酸是翻译后修饰的热点。Aβ 后修饰是一个广阔的但并未深入研究的领域,有可能对理解AD的发病机制做出重大贡献。酶切和修饰过程导致致病性Aβ 种类的形成,其水平可能随着年龄增加而增加,并和遗传因素相关。这些Aβ 的酶切和修饰可能是散发性AD的主要贡献者,靶向这些肽或相关的酶可以作为一种新的治疗机制或提供一种新的诊断方法。目前已经成功开发了多种检测Aβ 变体的方法,但没有一个黄金标准,为了实现对Aβ及Aβ变体的临床检测,还需要做大量的工作来开发简单、可靠、准确和便宜的技术。这些方法与用于生物传感的新纳米材料或探针的进一步整合将有助于获得高灵敏度和高选择性的Aβ 检测。对全长Aβ 的检测不足以获得关于AD 进展的明确诊断结论,通过高通量分析可以方便地对不同标志物进行多重检测。随着对AD发病机理的深入认识和纳米技术、设备制造方法的进步,应该会找到一种早期诊断AD的方法。
