全组份木质微球的制备及其工艺研究
2022-02-09邱良木马晓军
邱良木,刘 鑫,马晓军
(天津科技大学轻工科学与工程学院,天津 300222)
活性炭因其高比表面积、大孔容、丰富的表面官能团以及物理化学性质稳定等优点,被广泛应用于气体吸附[1-2]、超级电容器[3-4]、催化剂载体[5-6]、废水处理[7-8]、生物医药学等领域。作为近年来新兴的活性炭,球形活性炭具有良好的滚动性、耐磨性、延展性,且粒径易控、孔径易于分布,已逐渐成为研究热点。当前,球形活性炭的制备原料主要以沥青[10]、煤焦油[11]、酚醛树脂[12-13]、聚丙烯[14]等化石资源为主。受石油危机和环境保护等因素影响,开发新型球形活性炭材料已经备受关注。
木质生物质资源丰富且可再生,已经成为极具利用价值的潜在化工原材料[15]。近年来,尽管研究者利用木质资源组份开发出不同类型的木质碳球,但由于大多数木质生物质原料是天然高分子聚合物,且难熔和难溶,在木质碳球的制备过程中,其前驱体微球的制备只能利用部分组分如纤维素、木素等完成,对原料的浪费非常严重,生物质资源利用率极低;而且在分离植物组分的过程中也会对环境造成污染。
为此,本文以生物质液化技术获得的木材液化产物为原料,以聚乙烯醇为分散剂,采用乳液聚合法制备出全组份木质原球(FWOS),在经固化处理后获得全组分木质微球(FWM)。研究了酚/木质量比、PVA质量分数、HMTA添加量、搅拌速率等对微球形貌和结构的影响,以确定微球的最佳制备工艺。
1 实验
1.1 实验材料
杉木木粉(0.2 mm<粒径<0.8 mm),购自天津本地木材加工厂;苯酚、磷酸,分析纯,天津市江天化工技术股份有限公司;六次甲基四胺,分析纯,天津大学科威公司;无水乙醇、甲醛、盐酸,分析纯,天津市风船化学试剂科技有限公司;聚乙烯醇,分析纯,北京益利精细化学品有限公司。
1.2 全组分木质原球(FWOS)和全组分木质微球(FWM)的制备
取杉木木粉(W)、苯酚(P)和磷酸按一定比例混合后加入至500 mL的三口烧瓶中,搅拌,油浴加热至160℃,液化反应2 h后形成木材液化物。将液化物与六次甲基四胺(HMTA)、无水乙醇混合,室温搅拌1 h后得到前驱体混合溶液。所得混合溶液滴加到已预先加热至65℃的聚乙烯醇(PVA)水溶液中,设定一定的搅拌速度,加热至130℃并保持反应1.5 h。待反应体系冷却至室温后,将产物用去离子水反复洗涤、过滤后,置于真空干燥箱(温度50℃、真空度0.1 MPa)干燥12 h,即得FWOS。
将FWOS浸泡于质量分数为18.95%的盐酸和质量分数为18.95%的甲醛的混合溶液中,然后以5℃/min的升温速率由室温升至95℃并保温2 h。冷却至室温后,将产物过滤、水洗数次至中性后,置于真空干燥箱(温度50℃、真空度0.1 MPa)烘干,即得FWM。
1.3 测试与表征
采用FEI Apreo型场发射扫描电子显微镜(美国Thermo Fisher Scientific公司)于2 kV的电压下观察样品的形貌。采用Nicolet 6700型傅里叶红外光谱仪(美国Thermo Fisher Scientific公司)以4.0 cm-1的分辨率、500~4000 cm-1的扫描范围对样品的化学键及官能团进行表征。采用MASTERSIZER-3500型马尔文激光粒径分析仪(英国MALVERN公司)对样品的粒径进行测量;采用Q500型热重分析仪(美国TA仪器公司)对样品进行测试,分析其分解温度及质量损失,氮气流量50 mL/min,升温速率10℃/min,升温至500℃;采用MASTERSIZER-3500型马尔文粒径分析仪(英国MALVERN公司)对样品的球径进行测试,测量范围为1.00 μm~3 500.00 μm。
2 结果与讨论
2.1 工艺因素对木质微球粒径的影响
2.1.1 酚/木(P/W)质量比
酚/木质量比对木质微球粒径的影响见图1。从图1(a)粒径分布曲线可以看出:酚/木质量比为5∶1、6∶1和7∶1时,粒径分布均较为均一;其中酚/木质量比为7∶1时,粒径主要分布在11.2~58.9 μm(约80.81%);当酚/木质量比降至4∶1时,粒径主要集中在28~127 μm(约79.69%),且此时微球的中值粒径(D50)增大。这是由于反应物液体黏度过大,导致FWM间相互粘连,使得所制得的FWM尺寸不一。
由图1(b)可知:随着酚/木质量比降低,FWM粒径逐渐增大。这是因为随着游离苯酚含量的减少,原位生成的甲醛含量相对充足,可提供足够的羟甲基与酚羟基进行缩聚反应,反应程度加深,分子链明显增长,使得FWM粒径增大。同时,采用面积平均径和体积平均径的比值(即D(3,2)/D(4,3))来表征FWM的球形度,数值越大表明FWM越接近球形。结果显示,球形度随酚/木质量比减小而呈先增后降趋势,可能是因为苯酚含量降低使其黏度增加,反应聚合程度增强,FWM形态更加紧固,球体不易变形即被拉长、压扁;但体系黏度过大则引起FWM易发生团聚,粒径增大、球形度下降。

图1 不同酚/木质量比时FWM的粒径分布、粒径及球形度
2.1.2 PVA质量分数
不同PVA质量分数时FWM的粒径分布、粒径及球形度见图2。从图2(a)可看出:随PVA质量分数的增大,微球的粒径分布峰变窄,中值粒径逐渐左移,说明粒径分布更加均一,且平均粒径降低。从图2(b)可以看出:PVA质量分数为4%~10%时的平均粒径降低最为显著,几乎呈现线性减小趋势。当PVA质量分数为4%时,FWM最大粒径达253 μm,而当质量分数为12%时,最小粒径仅为12.1 μm。球形度曲线表明,当PVA质量分数小于8%时,微球的球形度随PVA质量分数增加而显著提高,而当PVA质量分数超过8%时,随着PVA质量分数的增加,球形度提高并不明显。因此,当酚/木质量比为6∶1、HMTA添加量为木材液化物质量的15%、搅拌速率为500 r/min时,PVA质量分数选10%较为合适。
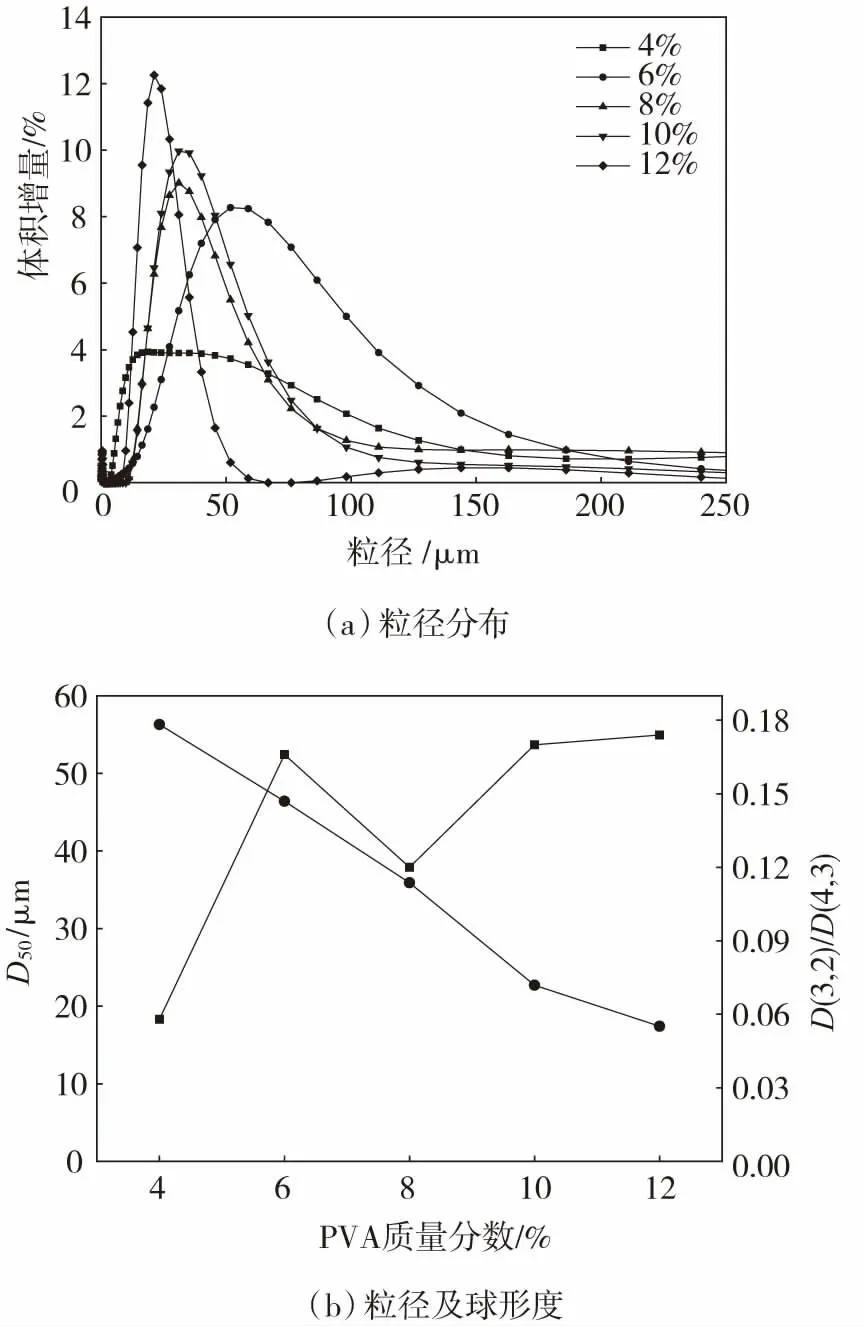
图2 不同PVA质量分数时FWM的粒径分布、粒径及球形度
2.1.3 HMTA添加量
不同HMTA添加量时FWM的粒径分布、粒径及球形度见图3。从图3可以看出:HMTA添加量对FWM的粒径没有太大影响。但其球形度呈现出先升后降趋势,在HMTA添加量为17.5%时显著降低,这是因为此时的FWM强度极低,易碎裂,难以保持球形状态。当HMTA添加量为7.5%时,由于反应结束后烧杯壁上存在大量黏稠的粘连物,使得制备的微球得率降低,球体间分散不均匀,粘连现象严重。这可能是因为固化剂添加量过少,使得反应时FWM得不到充分固化所致。当HMTA添加量为17.5%时,由于大量HMTA分解出的亚甲基桥(—CH2—)先与表层分子的活性位点互连,导致FWM内部的分子链增长受到一定的抑制,木质微球内外固化不均匀而破碎。
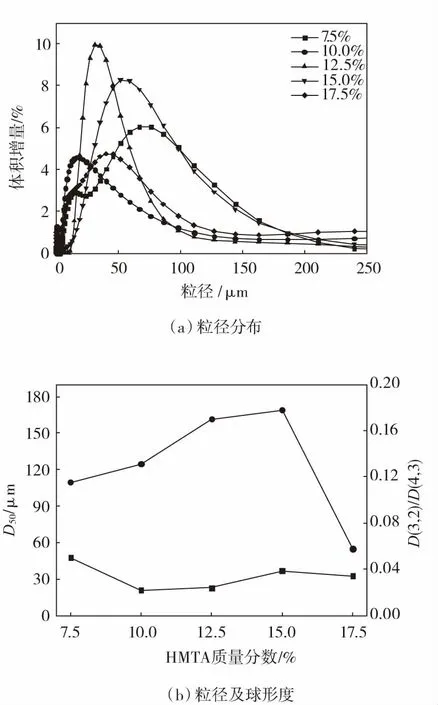
图3 不同HMTA添加量时FWM的粒径分布、粒径及球形度
2.1.4 搅拌速率
不同搅拌速率时FWM的粒径分布、粒径及球形度见图4。从图4(a)可以看出:随搅拌速率增加,曲线峰值向左移动且粒径区间逐渐变窄,说明FWM中值粒径呈减小趋势。350 r/min搅拌速率下出现双峰,表明在较低的搅拌速率下微球粒径分布不均一。搅拌速率为650 r/min时,粒径分布最集中且峰宽最窄,表明此时微球粒径的均一性最好。由图4(b)可看出:随着搅拌速率的增加,微球粒径先减小后增大。当搅拌速率低于650 r/min时,液滴的分散力占据主导地位,剪切力增强,粒径随转速增加而减小;而当搅拌速率超过“临界速率”650 r/min时,液滴中占据主导地位的聚并力大于分散力,此时粒径随转速增加而增大,其球形度也在达到“临界速率”后逐渐降低,这是因为原本搅拌分散开的FWM发生碰撞后又发生粘连现象,同时过大的粒径在动态搅拌下很难保持球状形态。

图4 不同搅拌速率时FWM的粒径分布、粒径及球形度
FWM的SEM图见图5。从图5(a)可以看出,较低的HMTA添加量使球体间分散不均匀,粘连现象严重。PVA质量分数为10%时获得的球体分散均匀(图5(b))。搅拌速率超过650 r/min时,过快的转速导致剪切力过强、液面张力增大,微球间发生碰撞聚并,部分形成类“花生”状球体(图5(c))。

图5 FWM的SEM图
综合考虑,酚/木质量比为6∶1,PVA质量分数为10%,HMTA添加量为木材液化物质量的15%,搅拌速率为650 r/min时制备出的全组分木质基微球形貌比较完整,粒径大小均匀。
2.2 固化前后木质基微球FTIR变化
FWOS和FWM的红外光谱图见图6。从图6可以看出,相比于FWOS来说,FWM在3467~3244 cm-1处羟基的特征吸收峰强度减弱[16],1358 cm-1处酚羟基的特征吸收峰几近消失,1226 cm-1处酚羟基的C—O特征吸收峰及1609 cm-1、1448 cm-1处苯环骨架振动吸收峰峰形均显著降低,表明在固化液强酸作用下,FWM中酚羟基之间、酚羟基与固化液中的+CH2OH碳正离子间发生了脱水缩合反应,分子间的交联能力得到提高[17]。2854 cm-1处出现强烈伸缩振动的亚甲基键,而1233cm-1次甲基醚键特征吸收峰大大减弱,说明FWM在固化处理时,大量次甲基醚键转化为亚甲基键,使FWM间主要靠热稳定性较好的亚甲基键通过酚羟基的邻位、对位方式相连接,形成网状交联结构,从而提高其力学强度[18]。1000~650 cm-1内多个C—H面外弯曲振动均发生相应变化,826 cm-1、753 cm-1处的特征吸收峰明显减弱,而879 cm-1处出现了新的特征吸收峰,同样表明FWM芳环与固化液中的+CH2OH碳正离子发生了交联反应,其内部形成了网状结构。

图6 FWOS和FWM的红外光谱图
2.3 固化前后木质基微球热重(TGA)分析
从图7中可看出,固化前后微球的TG曲线差异较大,具体可分为3个阶段:首先,从室温至160℃时,两样品均有轻微的质量损失,失重率均在7%之内,且FWOS的下降趋势更明显。在DTG曲线中,两样品的热失重速率变化基本一致,说明此阶段主要是大量游离苯酚和水分受热逸出,其中FWOS和FWM在69℃附近均出现峰,但FWM峰值明显低于FWOS,表明在此阶段FWOS失重更多。

图7 FWOS和FWM的TG和DTG曲线
其次,FWOS在160~360℃附近出现了强烈的热失重现象,其DTG曲线呈现出较明显的尖峰,峰温在260℃左右。FWM的DTG曲线峰高降低、峰形变宽,热解速率降低,峰温出现在410℃,较FWOS向高温方向偏移了150℃,表明FWM的分子交联程度提高,分子聚合程度增加,耐热性相应提高。
最后,从TG曲线可看出,440~800℃时两样品的热失重均减缓。FWOS在此阶段的热失重率较大,可能是因为在分子聚合过程中一些小分子链被断开,体系中未形成大分子长链网状结构,在此阶段大量酚类物质及CO2、CH4等小分子物质分解逸出,使得其在高温阶段的热失重较大。DTG曲线表明,FWM峰温右移了60℃左右,说明固化处理有利形成稳定的网状结构。从热解残余率可知,FWM比FWOS的残余率高(分别为55.34%和46%),同样表明经过固化工艺处理的FWM热稳定性较好。
3 结论
本文通过液化技术和乳液聚合法制得全组份木质基原球。当酚/木质量比为6∶1、PVA质量分数为10%、HMTA添加量为15%、搅拌速率为650 r/min时,所制得的微球形貌、结构较为理想。采用盐酸甲醛混合溶液固化处理可实现FWM强度和热稳定性的提高,为后续制备性能优良的全组分木质基炭球及其应用奠定了良好基础。
