1例罕见Melnick-Needles综合征患者基因变异分析
2022-02-02谢丹黄盛文吴江芬张文仪吴鹏安邦权
谢丹,黄盛文,吴江芬,张文仪,吴鹏,安邦权,4
(1.贵州大学医学院,贵阳 550025;2.贵州省人民医院产前诊断中心,贵阳 550002;3.遵义医学院,贵州遵义 564699;4.贵州省人民医院纪检监察室,贵阳 550002)
Melnick-Needles综合征(Melnick-Needles syndrome,MNS)又称骨结构不良综合征,是临床表型重叠的5种X连锁疾病中的一种,其余4种依次为耳腭指综合征Ⅰ型、耳腭指综合征Ⅱ型、额骨干骺端结构不良和终端骨发育不良,这些统称为耳腭指疾病谱(otopalatodigital syndrome spectrum disorders,OPDSD)[1-4]。迄今为止,全世界报道的MNS病例不到70例[4-5]。患有这种疾病的多数是女性,具有典型的特征性面容:眼球突出、脸颊丰满、小颌畸形,并伴有身材矮小、骨骼异常等[6]。因MNS极其罕见,且有些病例是致命的,大多数情况下,在家族中检测出来即为新发疾病,其以X连锁显性方式遗传[7-8]。本研究用全外显子测序和Sanger测序技术对1例临床疑似MNS患者进行临床表型和基因变异分析,明确其可能的致病原因,为患者提供适当的产前筛查策略和遗传咨询。
1 对象与方法
1.1研究对象 先证者,女,21岁,2021年3月于贵州省人民医院就诊。先证者1岁时开始出现胸廓发育异常,呈鸡胸和漏斗胸。8岁时出现脊柱后凸,双眼突出,生长发育迟缓,身材矮小,无智力障碍。近5年反复出现心悸、胸闷、咳嗽、咳痰、呼吸困难。查体:身高130 cm,体重25 kg;特殊面容:双眼突出,眼距增宽,上唇较厚,下巴短小;脊柱后突,胸廓畸形;手指和脚趾细长,末端粗大,呈爪型手。见图1。CT检查提示:鸡胸合并漏斗胸,双肺灌注异常。DR检查提示:颈椎生理曲度变直,脊柱胸段后突。心脏彩超检查提示:右房、右室增大,二尖瓣局限性返流,二尖瓣前瓣尖稍脱垂,微量反流。家族中无类似患者。
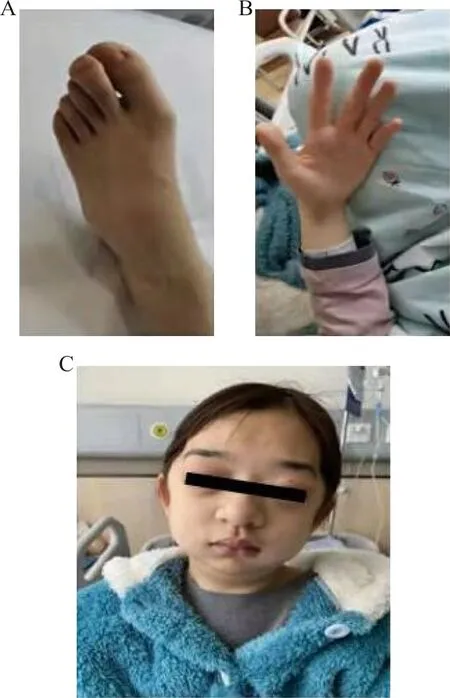
注:A、B,手指和脚趾细长,末端粗大;C,特殊面容,上唇较厚,下巴短小。图1 患者手脚部表现及特殊面容
1.2主要仪器与试剂 Nanodrop one超微量分光光度仪(美国赛默飞世尔公司);NovaSeq 6000基因测序仪(美国Illumina公司);ABI3730xl测序仪(美国Applied Biosystems公司); QIAamp DNA blood Mini kit(德国QIAGEN公司)。
1.3标本采集与处理 在征得先证者和家属的知情同意后,收集先证者及其父母的外周静脉血标本,用QIAamp DNA blood Mini kit 提取先证者及其父母全基因组DNA(操作按照试剂盒说明书进行),DNA 纯度和浓度用Nanodrop one超微量分光光度仪检测,DNA样品于-20 ℃或-80 ℃保存备用。本研究方案获得贵州省人民医院伦理学委员会批准[批准文号:伦审(科研)2022-05号]。
1.4全外显子组测序 将先证者及其父母DNA样品送至北京贝瑞和康医学检验实验室有限公司进行全外显子组高通量测序。首先从200 μL外周血中提取1 μg基因组DNA。50 ng DNA被裂解酶打断至200 bp左右。然后对DNA片段进行末端修复,并在3′端添加一个A碱基。用接头连接DNA片段,用XP微珠收集到约320 bp的片段。PCR扩增后,根据NanoWES说明书的步骤杂交并捕获DNA片段。对杂交产物进行洗脱和收集后进行PCR扩增和纯化。最后,用NovaSeq 6000 基因测序仪,采用150 bp的双端测序模式对该家族的基因组DNA进行测序。用Burrow-Wheeler比对工具将测序读数与人类参考基因组(HG19/GRCh37)进行比对。利用Verita Trekker®变异位点检测系统和Enliven®变异位点注释解读系统对数据进行分析。注释数据库主要包括: gnomAD(http://gnomad.broadinstitute.org/),人群千人基因组(http://browser.1000genomes.org), OMIM(http://www.omim.org),ClinVar(http://www.ncbi.nlm.nih.gov/clinv.ar),HGMD(http://www.hgmd.org),HPO(https://hpo.jax.org/app/)等。
1.5Sanger测序 根据GenBank (NM_001110556.2)中的参考序列设计PCR引物(F:GTGGTTCCCTGCT
TTGACG;R:CCGCCGTACTTGATGGTGA)。PCR反应体系为25 μL,包括 12.5 μL mix,10 μmol/L 上、下游引物各1 μL,ddH2O 9.5 μL。反应条件:95 ℃ 10 min;95 ℃ 30 s,60 ℃ 30 s,72 ℃ 30 s,共35个循环;72 ℃ 10 min。用PCR法进行扩增,以PCR扩增产物进行一代测序验证。用ABI3730xl测序仪进行Sanger测序,测序结果使用“Mutation Surveyor”软件与参考序列进行比对分析。
1.6生物信息学分析 HomoloGene系统分析错义变异的氨基酸位点是否具有跨种属保守性;根据美国医学遗传学与基因组学学会(American College of Medical Genetics and Genomics, ACMG)遗传变异分类标准与指南分析变异位点的致病性[9]。
2 结果
2.1全外显子组测序 先证者样本检测到FLNA基因的1个变异:NM_001110556.2: c.3562G>A(p.A1188T),导致FLNA蛋白第1 188位的丙氨酸替换为苏氨酸。判读为致病,与X连锁显性遗传疾病Melnick-Needles综合征(OMIM:309350)相关。
2.2Sanger测序验证 先证者在FLNA基因上发生c.3562G>A杂合突变,父亲和母亲均未携带该变异(图2),因此为新发变异。
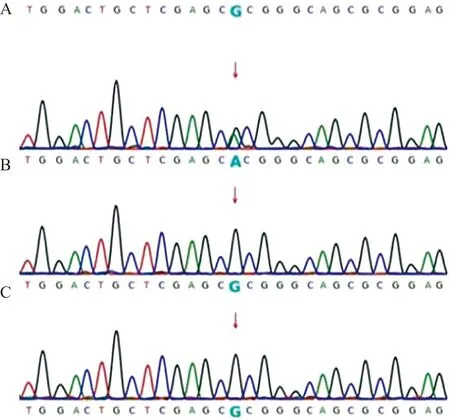
注:A,先证者FLNA基因的c.3562G>A变异;B,先证者父亲该位点为野生纯合型;C,先证者母亲该位点为野生纯合型。图2 Sanger测序结果
2.3变异位点致病性分析FLNA基因第22外显子存在c.3562G>A错义变异,根据Sanger测序结果,该变异只有先证者检出,父亲和母亲均未发生突变,故属于新发变异(PS2);参考gnomAD、人群千人基因组(1000G)等数据库,未见该变异的收录(PM2)。
根据文献报道,共在多例(6~14例)Melnick-Needles综合征疾病先证者中检出该变异[PMID:12612583;29575627] (PS4_Moderate); 经统计方法(REVEL)预测,结果显示该变异对基因或基因产物造成有害影响(PP3); 此外,该基因错义变异是造成相关疾病表型的常见机制,且良性错义变异比例低(PP2)。根据ACMG指南,该变异的证据为PS2、PM2、PS4_Moderate、PP3、PP2,故归类为致病性变异。
2.4生物信息学分析 经HomoGene和T-coffee系统对人、猕猴、家犬、牛、鼠、原鸡6个物种的FLNA基因氨基酸序列进行同源性多重比对,结果表明第1 188位丙氨酸(Ala)均高度保守(图3)。

图3 不同物种之间保守性分析
3 讨论
Melnick-Needles综合征是一种遗传性骨骼发育不良疾病[10]。1966年, Melnick和Needles首次描述了一些家庭成员患有严重的先天性骨骼疾病,这些疾病表现出典型的面部特征,如眼球突出、面颊饱满、小颌畸形,并伴有下肢S形弯曲、肋骨不规则收缩、颅底硬化[3]。本例先证者有部分表型符合以上描述,此外,还累及心脏、输尿管和肺部,虽然MNS的发病机制尚未确定,但泌尿系统、肺部和心脏受累在MNS先证者中也很常见[11-12]。
Robertson等[13]于2003年报道该综合征由编码细丝蛋白A(filamin A)的FLNA基因突变引起,此突变导致蛋白质的功能缺陷。FLNA是1个X连锁基因,位于Xq28,由48个外显子组成,其编码的蛋白质是一种广泛表达的蛋白质,通过与整合素、跨膜受体复合物和第二信使相互作用调节肌动蛋白细胞骨架的重组,在膜稳定性、交联F-肌动蛋白结合其他与细胞信号和细胞骨架完整性整合功能一致的蛋白质方面具有重要作用,该基因的缺陷是导致多种综合征的原因[14-17]。
MNS先证者表现出不同的表型,例如,Sheen等[18]报道了单卵双胞胎姐妹,但其中只有1个患MNS;又如Oh等[4]报道的病例中,尽管家族女性成员的表型严重程度不同,但她们都有相同的FLNA基因突变(c.3578 T>C,p.L1193P)。此外,对于男性来说表型的严重程度取决于母亲,未受影响母亲所生的男性表现出与女性相似的表型,受影响母亲所生的男性通常具有更为严重表型,表现为突眼、角膜硬化、高血压、严重的小颌畸形、腭裂、脐膨出、肾发育不良、梗阻性泌尿系统畸形、手脚畸形、颈胸前凸、不规则肋骨、长骨弯曲以及拇指发育不全或缺失[1,7]。因此, MNS先证者的表型以身材矮小和骨骼发育不良为主,可能因环境和遗传因素而略有不同。除此之外,如上所述,男性和女性的表型也因母亲是否受MNS影响而呈明显差异。
在临床诊断中,与MNS表型重叠的疾病除了耳腭指谱系疾病之外,还有粘多糖贮积症(mucopolysaccharidosis,MPS),其是一种因蛋白聚糖降解酶先天性缺陷所引起的蛋白聚糖分解代谢障碍性疾病;由于溶酶体内酶活性下降或者缺乏,机体分解粘多糖障碍,导致这些粘多糖分子在细胞内、血液和结缔组织中沉积,从而产生相应的临床症状,可分为MPSⅠ型、Ⅱ型、Ⅲ型、Ⅳ型、Ⅵ型、Ⅶ型、Ⅺ型和数种亚型[19]。在鉴别诊断中,MNS与MPS较显著的区别是MPSⅠ型、Ⅱ型、Ⅲ型、Ⅶ型和Ⅺ型表现出不同程度的智能落后,而Ⅳ型、Ⅵ型虽智力正常,但存在角膜混浊这一较典型的特征。本例先证者就曾误诊为MPSⅥ型。由于临床表现的多样性,在不同患者中还应根据实际情况进行分析鉴别。
目前国外已报道数十例MNS患者,国内也可见相关综合征的报道。本例先证者检出FLNA基因c.3562G>A错义变异,使得FLNA基因编码的蛋白第1 188位氨基酸由丙氨酸变异为苏氨酸(p.A1188T),从而导致多肽产物的氨基酸序列改变。文献报道的病例中FLNA基因的变异主要聚集在特定区域,如发生在外显子3~5、22、28~33和43~45[16]。而本例先证者变异位于第22外显子,是MNS变异的主要聚集区域。Sanger测序验证结果显示先证者携带的FLNA基因变异为新发,再发风险低,但不排除其亲代之一为生殖腺嵌合的可能。
综上所述,通过外显子组测序确诊了1例Melnick-Needles综合征患者。该病例丰富了MNS基因变异与临床表型谱,结合基因检测为MNS的分子诊断、遗传咨询和产前诊断提供了依据。
