1,2-双(二叔丁基膦甲基)苯的合成
2022-01-27王清福白东亚屈凤波刘婷婷
陈 辉,王清福, 周 铎,白东亚,张 垚,屈凤波,刘婷婷*
(1.河南省科学院 化学所有限公司,河南 郑州 450002; 2.河南中烟工业有限责任公司 技术中心,河南 郑州 450000)
0 引言
目前,有机双膦配体参与的过渡金属催化偶联反应广泛应用于C-C、C-N、C-O、C-H等化学键的构建中[1-6]。双膦配体的结构比较稳定,与中心金属通过双齿配位构造的反应环境单一,产物能获得较高的反应收率。双膦配体与过渡金属配位时,膦原子与中心金属的配位角度、膦原子上取代基的立体电子效应、空间位阻效应及配位骨架的刚柔性,很大程度上影响了双膦配体的催化反应活性[4]。目前所发展的双膦配体多以联萘或联苯类为主,这类配体在手性药物关键中间体的合成步骤上发挥了很大作用,具有实际的工业化应用价值[7-11]。
诸多广泛研究的双膦配体中,1,2-双(二叔丁基膦甲基)苯与金属催化剂共同作用,并应用于共轭二烯的氢甲酰化,C=C、C=N双键的加氢反应以及乙烯和一氧化碳的共聚反应,并取得了较好的反应结果[12-15]。然而,关于1,2-双(二叔丁基膦甲基)苯的合成文献中并未给出详细的报道。尽管SCHERER等[16]报道了一种通过季磷盐化生成1,2-双(二叔丁基膦甲基)苯双溴化氢盐,然后通过与三乙胺反应得到1,2-双(二叔丁基膦甲基)苯,但反应收率较低,只有10%。本文以简单易得的氯代叔丁烷为原料,通过格氏反应,将亚磷酸二乙酯转化为二叔丁基氧化膦,然后利用四氢铝锂作为还原剂,将二叔丁基氧化膦还原为二叔丁基膦氢;二叔丁基膦氢经正丁基锂锂化后,通过与邻二氯苄进行亲电反应,得到具有大位阻效应的有机双膦配体1,2-双(二叔丁基膦甲基)苯(见图1),经NMR核磁共振进行表征,确定了其结构。此外,本文还对关键步骤反应条件进行了优化。

图1 1,2-双(二叔丁基膦甲基)苯的合成线路Fig. 1 Synthetic routes of 1,2-bis(di-tert-butylphosphinomethyl)benzene
1 实验部分
1.1 仪器与试剂
WRR型目视熔点仪;Bruker Avance 400 MHz型超导核磁共振谱仪;STA-449C型元素分析仪。所用试剂均为化学纯。
1.2 合成
1.2.1 二叔丁基氧化膦(2)的合成
依据文献[17],在氩气保护下,向1500 mL的四口瓶中加入0.1 g碘作为引发剂,溶剂无水乙醚50 mL,并加入1 mol·L-1盐酸浸泡、丙酮洗涤后的镁条24 g(1.0 mol)。通过恒压滴液漏斗加入氯代叔丁烷92 g(1.0 mol)的乙醚溶液500 mL,滴加50 mL后,成功引发该反应,然后搅拌下继续滴加剩余氯代叔丁烷。滴加完成后,保持体系回流至镁条反应完全。停止反应,将体系用冰水浴冷却至0 ℃,滴加亚膦酸二乙酯55.2 g (0.4 mol),滴加完成后,在室温条件下反应15 h。再次将反应体系用冰水冷却至0 ℃,然后滴加1 mol·L-1盐酸溶液,并将反应体系pH值调节至6~7,继续搅拌反应2 h。停止反应,将反应体系静置后分液,有机相用水洗涤3次后,再用无水MgSO4干燥,过滤掉干燥剂后,减压除去乙醚,粗产物经硅胶柱层析(洗脱剂:V(乙酸乙酯)/V(正己烷)=1/1)纯化得白色固体二叔丁基氧化膦2 53.1 g,收率82%.1H NMR (CD2Cl2):δ6.17 (d,JP-H=449.8 Hz, 1 H, PH), 1.29 (d,J=16.0 Hz, 18 H) ;13C NMR (CD2Cl2):δ33.8 (d,J=58Hz), 25.4 (d,J= 2 Hz) 。31P NMR (CD2Cl2):δ72.4。
1.2.2 二叔丁基膦氢(3)的合成
依据文献[18],在氩气保护下,冰水浴冷却至0 ℃后,向三口瓶中加入无水乙醚10 mL,再加入LiAlH411.4 g (0.3 mol),搅拌下滴加二叔丁基氧化膦48.6 g (0.3 mol)的乙醚溶液50 mL,滴加完成后,在0 ℃条件下再反应0.5 h;将反应体系升至室温,再继续反应5 h。停止反应,将反应体系用冰浴降温至0 ℃,加入盐酸(1 mol·L-1)10 mL并淬灭反应,反应体系静置分层,将上层乙醚相在氩气保护下转移至另一氩气保护的三口瓶中,先加热蒸馏除去乙醚,然后继续加热蒸馏收集150~155 ℃馏分,得无色液体二叔丁基膦氢41.3 g,收率85%;1H NMR (CDCl3, 400 MHz)δ: 1.10 (d,J=191 Hz, 36H, C(CH3)3);31P NMR (CDCl3, 162 MHz)δ: -20.0 (J=191 Hz)。
1.2.3 1,2-双(二叔丁基膦甲基)苯(4)的合成
依据文献[16],在氩气保护下,向500 mL Schlenk瓶中加入二叔丁基膦氢(16.2 g,0.1 mol)的THF 溶液100 mL,将体系降低至-78 ℃后,缓慢滴加40 mL正丁基锂溶液(2.5 mol·L-1),滴加完成后,将反应体系升至室温,继续反应1 h;然后再缓慢滴加邻二氯苄(8.8 g 0.05 mol)的THF溶液10 mL,滴加完成后,反应过夜。停止反应,向反应体系中滴加40%四氟硼酸水溶液330 g,体系中生成大量固体。当滴加完成后,在室温条件下搅拌1 h,将反应体系抽滤,所得产物固体用二氯甲烷/正己烷重结晶,真空干燥后得白色固体1,2-双(二叔丁基膦甲基)苯27.6 g,收率70%。1H NMR(CDCl3, 400 MHz)δ: 1.14 (d,J=10.8 Hz, 36H), 3.04 (d,J=2.8 Hz, 4H), 7.05~7.07 (m, 2H), 7.52~7.55 (m, 2H);13C{1H} NMR(CDCl3, 100 MHz)δ: 26.4 (dd,J=24.0 Hz,J=4.7 Hz), 29.9 (d,J=13.1 Hz), 31.9 (d,J=22.5 Hz), 125.2 (d,J=1.8 Hz), 138.7 (dd,J=15.1 Hz), 138.7 (dd,J=9.4 Hz,J=2.6 Hz)。31P{1H} NMR(CDCl3, 162 MHz)δ: 28.5。
1.2.4 钯氢制备
1,2-双(二叔丁基膦甲基)苯(dtbpx)与Pd(OTf)2以1∶1的摩尔比在甲苯中回流反应8 h,将所得固体抽滤后重结晶,制备[(dtbpx)Pd(OTf)2]。将10 mg [(dtbpx)Pd(OTf)2] 以及3当量(dtbpxH2)(OTf)2混合,并加入0.5 mL CD2Cl2及0.3 mL甲醇,经核磁表征分析,生成钯氢物种。dtbpx为1,2-双(二叔丁基膦甲基)苯。
1,19-二酯:1H NMR(CDCl3, 400 MHz)δ: 1.21~1.35 (m, 26H), 1.58~1.71 (m, 4H) , 2.35 (t,J=7.56 Hz, 4H), 3.72 (s, 6H);13C NMR (100 MHz, CDCl3, 298 K)δ: 25.0, 29.3, 29.4, 29.5, 29.7, 34.3, 51.6, 174.8。[19]
2 结果与讨论
2.1 二叔丁基氧化膦的合成工艺分析
在二叔丁基氧化膦的合成中,首先利用金属镁与氯代叔丁烷进行格氏化反应,再经与亚膦酸二乙酯反应生成二叔丁基氧化膦。然后对影响该反应收率的条件进行优化,结果如表1所示。
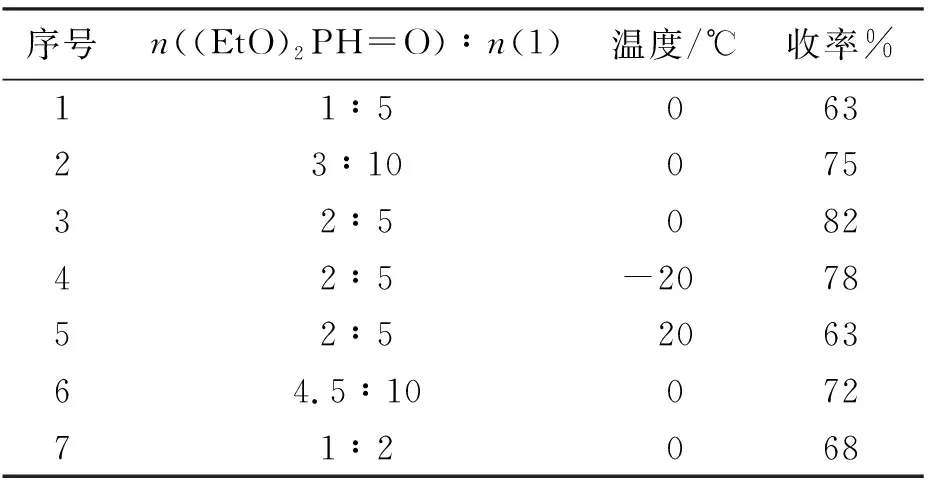
表1 亚膦酸二乙酯的量及反应温度对反应的影响Tab. 1 Effect of the amount of material and temperature on the reaction
首先,考察了亚膦酸二乙酯的反应用量对二叔丁基氧化膦收率的影响。在0 ℃反应条件下,当n(亚膦酸二乙酯)∶n(氯代叔丁烷)=1∶5时,二叔丁基氧化膦收率为63%。然后增加亚膦酸二乙酯用量,当n(亚膦酸二乙酯)∶n(氯代叔丁烷)=3∶10时,二叔丁基氧化膦收率为75%。当n(亚膦酸二乙酯)∶n(氯代叔丁烷)=2∶5时,二叔丁基氧化膦收率达到最高82%。当继续增加亚膦酸二乙酯的用量,二叔丁基氧化膦收率反而逐渐降低,这可能是由于单取代产物的生成,造成双取代产物的减少。同时,在n(亚膦酸二乙酯)∶n(氯代叔丁烷)=2∶5的条件下,还考察了体系反应温度对二叔丁基氧化膦收率的影响。当体系反应温度为0 ℃,二叔丁基氧化膦收率为82%,将体系反应温度升至20 ℃时,二叔丁基氧化膦收率降至63%;当将反应温度降至-20 ℃时,二叔丁基氧化膦收率也略微降低,为78%。因此,最终选择最佳反应温度为0 ℃。
2.2 1,2-双(二叔丁基膦甲基)苯的合成工艺分析
在化合物1,2-双(二叔丁基膦甲基)苯的合成中,对反应溶剂进行了筛选。当反应溶剂为乙醚时,1,2-双(二叔丁基膦甲基)苯的收率为58%;以四氢呋喃作为反应溶剂时,1,2-双(二叔丁基膦甲基)苯4收率为62%。但溶剂为正己烷或甲苯时,1,2-双(二叔丁基膦甲基)苯的反应收率降低为5%及35%,所以选择四氢呋喃为反应溶剂。
2.3 1,2-双(二叔丁基膦甲基)苯的催化活性
为了验证生成配体的催化活性,将以上得到的双磷配体与金属钯配位得到1,2-双(二叔丁基膦甲基)苯的钯氢配合物(见图2),其在油酸甲酯的甲氧基羰基异构化反应中表现出优异的催化活性,对线性产物1,19-二酯化合物的选择性大于90%[13]。同时,向Pd前体中添加略过量配体的三氟磺酸盐,对油酸甲酯的甲氧基羰基异构化反应的催化活性可以提高6倍以上。
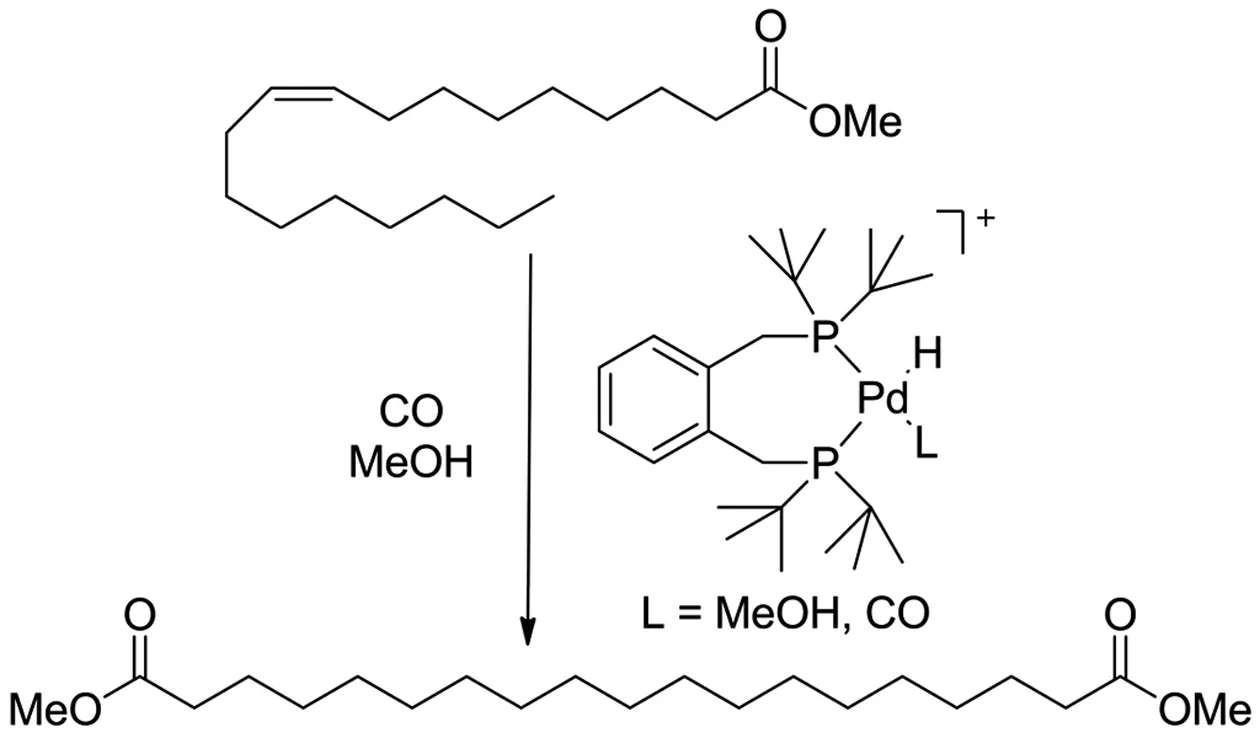
图2 1,2-双(二叔丁基膦甲基)苯的催化活性Fig. 2 Catalytic activity of 1,2-bis(di-tert-butylphosphinomethyl)benzene
3 结论
以氯代叔丁烷为起始原料,通过格氏反应,将亚磷酸二乙酯转化为二叔丁基氧化膦,然后利用四氢铝锂作为还原剂,将二叔丁基氧化膦还原为二叔丁基膦氢;二叔丁基膦氢经正丁基锂锂化后,通过与邻二氯苄进行亲电反应,得到具有大位阻效应的有机双膦配体1,2-双(二叔丁基膦甲基)苯,并对关键反应步骤进行了条件筛选。通过对1,2-双(二叔丁基膦甲基)苯合成的深入研究,可以提高有机膦的理论水平,促进新型高效有机膦催化剂的产生,为药物产业的发展做出贡献。
