柱前衍生高效液相色谱法测定尼莫地平注射液中的甲醛和乙醛
2022-01-27顾晓风杨袁郝刚唐倩倩
顾晓风,杨袁,郝刚,唐倩倩
(1.苏州市药品检验检测研究中心,江苏苏州 215104;2.山东绿叶制药有限公司,山东烟台 264670)
尼莫地平为第2 代二氢吡啶类Ca2+拮抗剂,其注射剂是预防和治疗动脉瘤性蛛网膜下腔出血后脑血管痉挛引起的缺血性神经损伤的首选药[1–2]。尼莫地平为难溶性药物,处方中可通过添加聚乙二醇400 提高尼莫地平的溶解性。聚乙二醇为环氧乙烷和水缩聚而成的混合物,具有以氧乙烯基为重复单位组成的线性链状结构,常作为助溶剂、稳定剂、乳化剂应用于药物制剂中,属于高风险性药用辅料[3]。《中国药典》定义药用辅料为生产药品和调配处方时使用的赋形剂和附加剂,与药物的不相容性表现在其本身与药物的反应及辅料中的杂质与药物的反应,可能对制剂的质量、安全性和有效性产生影响[4–5]。聚乙二醇类药用辅料为聚氧乙烯的大分子聚合物,受光照或金属离子催化时会发生自氧化反应,生成甲醛和乙醛等小分子杂质[6],这些杂质可能与尼莫地平中的敏感官能团发生缩合反应,引发主药的降解;同时,甲醛和乙醛均被世界卫生组织公布为1 级致癌物质,在体内会增加氧化应激和氧化损伤,引起基因突变、染色体损伤,甚至引起严重的染色体重排,导致各种血癌[7]。按照自氧化反应的机理,制成制剂后,聚乙二醇类药用辅料的自氧化反应并不会停止,而随着贮存时间的延长,制剂中甲醛和乙醛含量可能会比辅料中含量有所提高,故控制制剂中甲醛和乙醛的含量,对有效控制高风险制剂的安全性有重要意义。
《中国药典》2020 年版采用紫外衍生化法测定聚乙二醇类药用辅料中甲醛的含量,对乙醛未作控制[3]。添加了聚乙二醇类药用辅料的制剂,对其中降解产物甲醛和乙醛的测定未见报道。测定原料药、药用辅料、食品等中甲醛含量的方法有分光光度法[8–9]、气相色谱法[10–11]、液相色谱法[12–15]、离子色谱法[16]等。分光光度法易受其它杂质如烯烃等物质的干扰,专属性不强;离子色谱法仪器昂贵,普及性不如气相和液相色谱法。
笔者尝试用顶空气相色谱法测定尼莫地平注射液中甲醛和乙醛的含量,结果表明乙醛色谱响应较差,方法灵敏度达不到要求,故采用2,4-二硝基苯肼(DNPH)进行柱前衍生化后以高效液相色谱法测定,该方法灵敏度高,操作简便,准确度好。
1 实验部分
1.1 主要仪器与试剂
高效液相色谱仪:LC–2010CHT 型,附带LC Solution 工作站,日本岛津公司。
电子天平:XS205DU 型,感量为0.1 mg,瑞士梅特勒–托利多公司。
乙腈:色谱纯,霍尼韦尔贸易(上海)有限公司。
2,4-二硝基苯肼:分析纯,上海麦克林生化科技有限公司。
盐酸:优级纯,中国医药集团有限公司。
乙醇:色谱纯,德国默克股份两合公司。
枸橼酸、枸橼酸钠:均为分析纯,中国医药集团有限公司。
甲醛水溶液标准物质:100 μg/mL,标准物质编号为BW 20040–100–W–20,国家标准物质中心。
乙醛对照品:质量分数为99.3%,中国食品药品检定研究院。
尼莫地平对照品:质量分数为99.7%,中国食品药品检定研究院。
尼莫地平注射液样品:规格为10 mg(50 mL),德国拜耳医药保健有限公司。
实验用水为去离子水,由密理博纯水仪制得。
1.2 色谱条件
色谱柱:Waters XBridge C8柱(250 mm×4.6 mm,5 μm);流动相:乙腈–水(体积比为50∶50),流量为1.0 mL/min;柱温:30 ℃;检测波长:360 nm;进样体积:20 μL。
1.3 溶液配制
1.3.1 衍生化溶液
取2,4-二硝基苯肼0.2 g,置于50 mL 棕色容量瓶中,加入乙腈约40 mL 及盐酸3 mL,超声10 min 使2,4-二硝基苯肼溶解,放冷,用乙腈稀释至标线。临用新制。
1.3.2 甲醛和乙醛混合对照品储备液
取甲醛水溶液标准物质和乙醛对照品适量,用水稀释成质量浓度分别为10.0,12.0 μg/mL 的甲醛和乙醛混合对照品储备液。
1.3.3 甲醛、乙醛系列混合标准工作溶液
精密移取甲醛和乙醛混合对照品储备液0.01、0.02、0.05、0.1、0.5、1 mL,分别 置于10 mL 容 量瓶中,加入乙腈1 mL,摇匀,再加入1 mL 衍生化溶液,摇匀,置于暗处,于室温下反应30 min,用乙腈稀释至标线,摇匀,制得甲醛质量浓度分别为0.01、0.02、0.05、0.1、0.5、1 μg/mL,乙醛质量浓度分别为0.012、0.024、0.06、0.12、0.6、1.2 μg/mL 的系列混合标准工作溶液。
1.3.4 样品溶液
精密移取尼莫地平注射液2 mL,置于10 mL 容量瓶中,加入乙腈1 mL,摇匀,再加入1 mL 衍生化溶液,摇匀,置于暗处,于室温下反应30 min,用乙腈稀释至标线,摇匀。
1.3.5 空白对照溶液
按处方取尼莫地平、乙醇、枸橼酸、枸橼酸钠,配制不含有聚乙二醇400 的空白溶液。取空白溶液2 mL,置于10 mL 容量瓶中,按1.3.4 方法进行衍生化,作为空白对照溶液。
1.4 实验方法
取甲醛和乙醛系列混合标准工作溶液及样品溶液,按1.2 色谱条件进样分析,记录色谱峰面积,建立甲醛和乙醛的标准工作曲线,计算线性方程,利用标准曲线法计算样品中甲醛和乙醛的含量。
2 结果与讨论
2.1 衍生化条件的选择
由于缺少发色基团,甲醛和乙醛需借助衍生化试剂生成衍生化产物。目前最常用的紫外衍生化试剂是2,4-二硝基苯肼(DHPH)。在偏酸性条件下,DHPH 与甲醛、乙醛反应,分别生成甲醛–2,4-二硝基苯腙(甲醛–DNPH)和乙醛–2,4-二硝基苯腙(乙醛–DNPH),该类化合物于350~380 nm 波长处有强紫外吸收,结合高效液相色谱分离技术,可建立具有灵敏度高、专属性强等优点的检测方法。
分别使用0.5、1、2 mL 衍生化溶液,考察衍生化试剂用量对测定结果的影响。结果表明,使用1 mL衍生化溶液所得色谱峰面积大于使用0.5 mL 衍生化溶液样品的色谱峰面积,而与使用2 mL 衍生化溶液相比,样品色谱峰面积无显著性差异,故选择使用1 mL 衍生化溶液.
分别将样品置于室温和45 ℃恒温水浴中反应30 min 和45 min,考察衍生化反应温度与时间对测定结果的影响,结果表明于室温反应30 min,即可反应完全,故确定衍生化条件为加入1 mL 衍生化溶液,置于暗处室温下反应30 min。
2.2 色谱条件的选择
2.2.1 检测波长
衍生化产物甲醛–DNPH 的乙腈溶液在350 nm波长处有最大紫外吸收,乙醛–DNPH 的乙腈溶液则在360 nm 波长处有最大紫外吸收,试验结果表明两个波长下样品的测定结果无明显差异,结合文献[7],选择检测波长为360 nm。
2.2.2 色谱柱
参考文献[7][14]分别考察C8和C18色谱柱,试验结果表明,常见的C8和C18色谱柱均能将目标物完全分开,而C18色谱柱目标物峰保留时间较长,拖尾因子较C8色谱柱大,故选择C8柱。比较了Agilent zorbax C8和Waters XBridge C8色谱柱,后者色谱峰形更加对称尖锐,故选择Waters XBridge C8柱(250 mm×4.6 mm,5 μm)作为实验用色谱柱。
2.2.3 流动相
文献报道多以乙腈–水系统为流动相测定甲醛–DNPH 和乙醛–DNPH。分别考察不同配比的乙腈与水作为流动相的分离分析效果,结果表明当乙腈与水体积比为65∶35 时,两个目标峰在5 min内出峰,甲醛–DNPH 峰与衍生化试剂峰分离度仅为1.5;当乙腈–水体积比为35∶65 时,甲醛–DNPH峰和乙醛–DNPH 峰的保留时间分别为16.4 min和24.4 min,峰形明显展宽;当乙腈体积分数为45%~55%时,均能得到峰形对称、分离度良好的色谱峰,故选择乙腈与水的体积比为50∶50。
2.2.4 柱温
分别考察柱温30、35、40℃对测定结果的影响,结果表明除保留时间略有不同外,测定结果无明显差异,故选择柱温为30 ℃。
2.3 系统适用性试验
精密量取空白对照溶液、甲醛质量浓度为0.1 μg/mL 和乙醛质量浓度为0.12 μg/mL 的混合标准工作溶液、样品溶液各20 μL,分别注入色谱仪,按1.2 色谱条件测定,色谱图如图1 所示。结果表明,衍生化产物甲醛–DNPH 峰和乙醛–DNPH 峰的理论板数均大于10 000,拖尾因子小于1.2,分离度大于5.0。尼莫地平及其它物质对甲醛–DNPH 峰和乙醛–DNPH 峰的测定均无干扰。
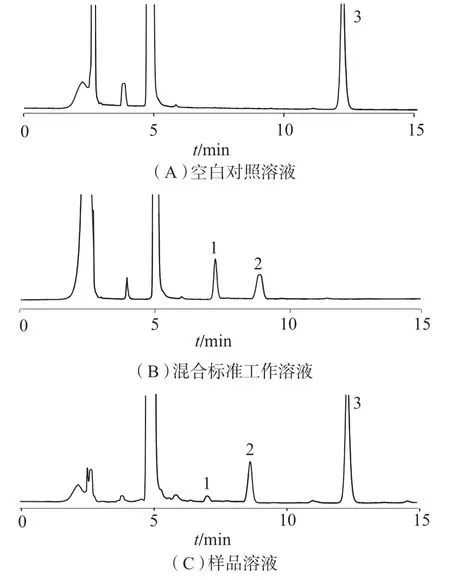
图1 高效液相色谱图
2.4 线性关系和检出限
取甲醛、乙醛系列混合标准工作溶液,按照质量浓度从低到高依次进样分析,记录色谱图,以质量浓度X(μg/mL)为自变量、色谱峰面积Y为因变量进行线性回归,得回归方程和相关系数。
取甲醛质量浓度为0.1 μg/mL 和乙醛质量浓度为0.12 μg/mL 的混合标准工作溶液,逐级稀释,测定并记录色谱峰面积。以信噪比3∶1 对应的质量浓度作为检出限。
甲醛、乙醛的质量浓度线性范围、线性方程、相关系数和检出限列于表1。

表1 甲醛、乙醛的质量浓度线性范围、线性方程、相关系数及检出限
由表1 可知,甲醛、乙醛的质量浓度分别在0.01~1.00 μg/mL 及0.01~1.20 μg/mL 范围与色谱峰面积具有良好的线性关系,线性相关系数均为0.999 9。该法测定甲醛、乙醛的检出限分别为0.003、0.004 μg/mL。
2.5 精密度试验
取同一批号样品6 份,按1.3.4 方法制备样品溶液,进样测定后记录色谱图,用色谱峰面积标准曲线法定量,甲醛、乙醛的质量浓度测定结果见表2。由表2 可知,甲醛、乙醛测定结果的相对标准偏差分别为0.86%、1.02%,表明本方法测量精密度良好,满足分析要求。

表2 精密度试验结果
2.6 加标回收试验
取甲醛、乙醛质量浓度分别为0.1、1.5 μg/mL的混合标准溶液,精密移取样品1.0 mL,置于10 mL 容量瓶中,平行制备9 份,每3 份一组,每组分别精密加入上述甲醛和乙醛混合标准溶液0.6、1.0、1.4 mL,再加入1 mL 衍生化溶液,摇匀,置于暗处于室温下反应30 min,用乙腈稀释至标线,摇匀后进样测定,计算回收率,结果列于表3。由表3 可知,3 个加标水平甲醛、乙醛的平均加标回收率分别为104.3%、92.0%,表明本方法测量准确度较高,满足分析要求。
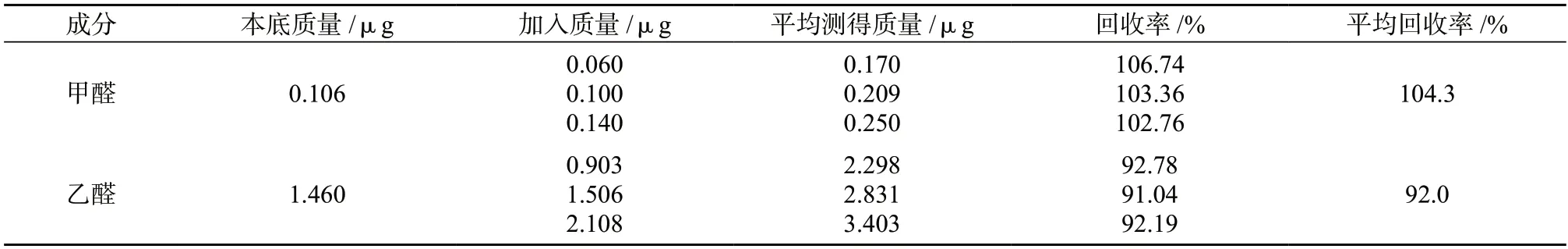
表3 加标回收试验结果
3 结语
建立了柱前衍生化HPLC 法,对尼莫地平注射液中的甲醛、乙醛进行测定,方法操作简单,灵敏度高,准确度高。该法有助于指导生产企业选择更优质的药用辅料,以提高药物的安全性。
