液相色谱特征图谱用于草乌成分定量
2022-01-27蒙天琛李鑫隆海鸣
蒙天琛,李鑫,隆海鸣
(哈尔滨商业大学药学院,哈尔滨 150076)
草乌为毛茛科植物北乌头(Aconitum kusnezoffii Reichb)的干燥块根。《中国植物志》对其有详细记载,我国的草乌药材资源十分丰富,大约有200 多个品种,在东北、河北省、内蒙古、山西省等地区分布较为集中。草乌药材的使用来源于古籍《神农本草经》,可以用于风寒湿痹的治疗等,已被广泛应用于临床[1-9]。部分药理学研究发现乌头类药物、复方制剂都有消除炎症、减轻疼痛和免疫抑制的作用。临床上多用于治疗各种各样的痛症,例如风湿痹痛等[10-15]。分析草乌的组成成分更有利于它在制剂中被有效应用。采用高效液相色谱(HPLC)法测定制草乌药材中的生物碱已有报道,但针对不同产地的草乌特征图谱以及指标性成分的含量测定尚未有报道。
笔者采用HPLC 法建立10 批不同地区草乌药材的高效液相指纹图谱,对样品进行相似性对比分析,并比较其生物碱含量的异同,考察不同产地草乌之间的质量差异,可以全面精确地为草乌药材的质量评价提供依据。
1 实验部分
1.1 主要仪器与试剂
高效液相色谱仪:Dionex UltiMate 3000 型,美国Dionex 公司。
超声波清洗机:AS3120A 型,深圳市洁盟清洗设备有限公司。
电子天平:FA22048 型,感量为0.1 mg,上海精科天美科学仪器有限公司。
氨水、甲醇:均为色谱纯。
新乌头碱对照品:批号为wkq20031307,质量分数不小于98%,四川省维克奇生物科技有限公司。
乌头碱对照品:批号为wkq20041408,质量分数不小于98%,四川省维克奇生物科技有限公司。
次乌头碱对照品:批号为wkq20031308,含量不小于98%,四川省维克奇生物科技有限公司。
草乌样品:10 批,经哈尔滨商业大学金哲雄教授鉴定为毛茛科植物北乌头(Aconitum kusnezoffii Reichb)的干燥块根,药材来源见表1。

表1 草乌样品产地
实验所用其它试剂均为分析纯。
实验用水为纯净水杭州娃哈哈集团有限公司。
1.2 色谱条件
色谱柱:WelchromC18柱(250mm×4.6mm,5μL,上海月旭科技有限公司);流动相:甲醇–0.1%三乙胺水溶液(体积比为75∶25),流量为1mL/min;检测波长:235 nm;柱温:30 ℃;进样体积:20 μL。
1.3 溶液配制
新乌头碱、乌头碱、次乌头碱对照品溶液:精密称取适量的新乌头碱、乌头碱、次乌头碱对照品,用甲醇配制成质量浓度分别为1.04、1.14、1.04 mg/mL的对照品储备液,分别取适量体积的新乌头碱、乌头碱、次乌头碱对照品储备液,配制成新乌头碱、乌头碱、次乌头碱质量浓度分别为52、28.5、26 μg/mL的混合标准溶液,冷藏备用。
草乌样品溶液:精密称取草乌粉末2.2 g,置于具塞锥形瓶中,加入35 mL 质量分数为0.04%的盐酸水溶液,搅拌6 h,以4 000 r/min 转速离心15 min,用质量分数为10%的氨水将上层清液pH 调至10,再向其中加入20 mL 乙醚进行萃取,取乙醚层,挥干乙醚,用甲醇溶解,并于25 mL 容量瓶中定容至标线,经0.45 μm 微孔滤膜过滤。
2 结果与讨论
2.1 色谱条件的优化
分别选择色谱柱WelchromC18柱(250 mm×4.6 mm,5 μL)和Agilent HILIC 柱(100 mm×2.1 mm,2.7 μL),结果显示前者对草乌样品溶液中各成分的分离效果好,能实现主要色谱峰的分离且色谱峰信息较全。
考虑乙腈毒性问题,选择常用流动相甲醇–水进行考察,结果显示基线稳定,分离度良好。但色谱峰形因草乌中的生物碱存在色谱峰拖尾现象,所以在水相中加入0.1%三乙胺溶液调整色谱峰形。分别改变流动相中有机相和水相的体积比为85∶15、75∶25、65∶35,考察流动相中有机相和与水相的配比对色谱分离效果的影响。结果显示在流动相中有机相和与水相的配比为75∶25 的条件下能够显示稳定峰高的色谱峰且分离度理想,故选择甲醇–0.1%三乙胺水溶液(75∶25)为流动相。
分别选择进样体积为10、20 μL,考察进样体积对色谱分离效果的影响,结果显示进样体积为10 μL 时色谱图中未出现目标色谱峰,故选择进样体积为20 μL。
分别设定柱温为20、30、40 ℃,考察不同柱温条件下草乌样品的色谱分离情况,结果显示柱温为30℃和40 ℃时色谱分离度较好,色谱峰宽较窄且基线稳定。考虑高温对色谱柱的寿命影响,故选择柱温为30 ℃。
2.2 草乌HPLC 特征图谱
2.2.1 色谱峰重复性
将草乌样品溶液连续测定6 次,其10 个共有色谱峰的面积列于表2。由表2 可知,草乌样品溶液其共有色谱峰面积测定值的相对标准偏差均小于2%,说明色谱峰重复性良好。

表2 草乌样品共有峰色谱峰重复性试验结果
2.2.2 样品稳定性
制备草乌样品溶液,分别放置0,2,4,8,12,24 h后进样测定,10 个共有色谱峰的面积列于表3。由表3 可知,草乌样品溶液共有色谱峰面积均值的相对标准偏差均小于2%,说明样品溶液稳定性良好。

表3 草乌样品溶液稳定性试验结果
2.2.3 色谱峰重现性
按照1.3 方法制备得到6 个草乌样品溶液,分别进行测定,所得共有色谱峰面积列于表4。由表4 可知,草乌样品溶液共有色谱峰面积测定值的相对标准偏差小于2%,表明样品测定色谱峰重现性较好。
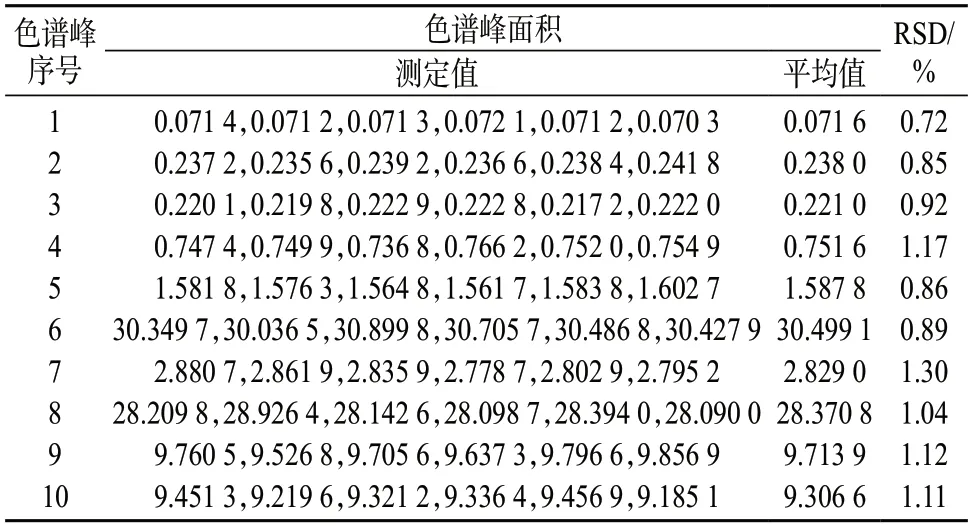
表4 草乌样品共有峰色谱峰重现性试验结果
2.2.4 特征峰鉴别
分别取新乌头碱、乌头碱、次乌头碱混合标准溶液、草乌样品溶液20 μL 进行测定,色谱图分别如图1、图2 所示。

图1 新乌头碱、乌头碱、次乌头碱混合标准溶液色谱图

图2 草乌样品溶液色谱图
分别取10 个不同产地的草乌样品,按照1.3 方法制备样品溶液并进行测定,用国家药典委员会的“中药色谱指纹图谱相似度评价系统2004A 版”对色谱图进行分析,以平均数法作为对照指纹图谱的生成方法,参照草乌样品溶液色谱图,标定了10 个共有峰,其中8、9、10 号峰分别为新乌头碱、乌头碱、次乌头碱的特征峰,如图3所示。将样品与参照图谱进行比较,计算其指纹图谱的相似度值,相关数据列于表5。由表5 可知:10 个草乌样品的液相色谱图与对照指纹图谱的相似度平均值均大于0.90,表明10 个产地的草乌品质均一。

表5 草乌样品指纹图谱相似度数据
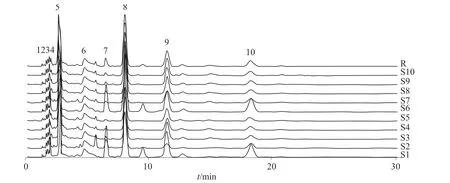
图3 10 个草乌样品液相色谱图
2.3 含量测定
2.3.1 线性关系
分别精密移取新乌头碱、乌头碱、次乌头碱对照品溶液各0.05、2.0、5.0、8.0 mL 于10 mL 容量瓶中,用甲醇稀释至标线,摇匀,配制成分别含有新乌头碱312.0、249.6、156.0、62.4、1.56 μg/mL,乌头碱36.75、29.4、18.38、7.35、0.18 μg/mL,次乌头碱26.0、20.8、13.0、5.2、0.13 μg/mL 的混合对照品溶液,经0.45 μm 微孔滤膜过滤后测定,以色谱峰面积为纵坐标(Y)、质量浓度为横坐标(X)进行线性回归。新乌头碱、乌头碱和次乌头碱的线性范围、线性方程、相关系数见表6。由表6 数据可知,3 种成分线性相关系数均大于0.999,表明线性关系良好,适于准确定量。
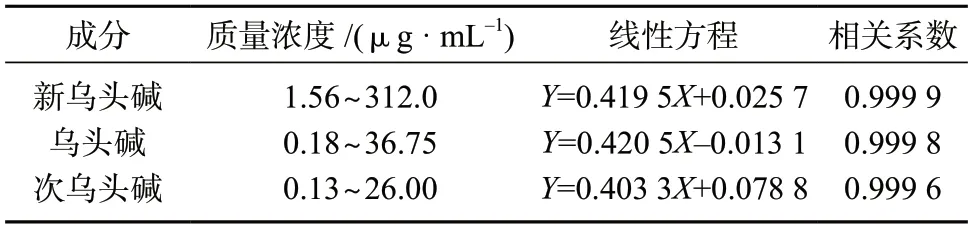
表6 新乌头碱、乌头碱、次乌头碱线性关系
2.3.2 重复性精密度
称取6 份草乌药材,按1.3 方法制备样品溶液,进样测定色谱峰面积,计算测定均值的相对标准偏差,结果列于表7。由表7 数据可知,各指标成分色谱峰面积的相对标准偏差均小于2%,表明该方法的重复性精密度良好。
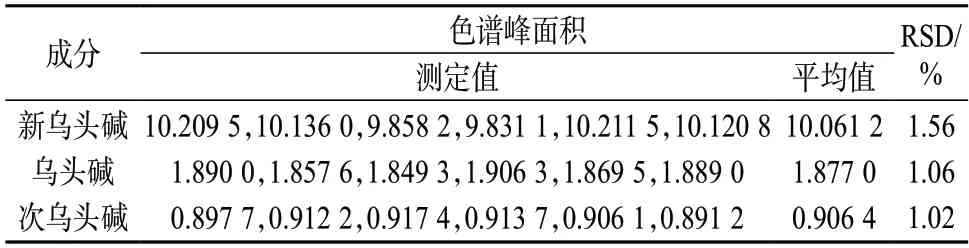
表7 重复性精密度试验结果
2.3.3 重现性精密度
精密吸取草乌样品溶液,利用本法在同日内测定3 次,并且在3 天内连续测定,计算日内、日间色谱峰面积测量值的相对标准偏差,试验结果列于表8。由表8 数据可知,新乌头碱、乌头碱、次乌头碱色谱峰面积的相对标准偏差均小于2%,表明方法的重现性精密度良好。

表8 重现性精密度试验结果
2.4 加标回收试验
选取草乌药材6 份,配制样品溶液后测定其中新乌头碱、乌头碱、次乌头碱的含量,分别向样品中加入新乌头碱、乌头碱、次乌头碱对照品适量,按照1.3 方法制备样品溶液,测定3 种组分的色谱峰面积,计算加标回收率,试验结果列于表9。

表9 加标回收试验结果
由表9 可知,3 种成分平均加标回收率为100.51%~100.66%,表明该方法测量准确度较高。
2.5 样品分析
取10 个不同产地的草乌粉末各约2.2 g,按1.3方法制备样品溶液,测定新乌头碱、乌头碱、次乌头碱的含量,实验结果列于表10。由表10 数据可知,产地1 的草乌的生物碱含量最高为3.404 6 mg/g,其余产地草乌的生物碱含量较为接近,并且10 个产地的草乌符合《中国药典》2020 年版关于草乌中生物碱总含量应为1.5~7.5 mg/g 的规定。

表10 不同产地草乌生物碱质量分数测定结果 mg/g
3 结语
采用HPLC 指纹图谱方法标定出草乌中的10个共有峰,指认出3 个特征峰,对草乌原药材进行了指纹图谱相似度评价,10 个草乌样品的HPLC 色谱图与对照指纹图谱相似度平均值均大于0.90,表明其相似度较好。应用所建方法测定了10 批不同产地的草乌中新乌头碱、乌头碱、次乌头碱3 种成分的含量。所建立的HPLC 指纹图谱方法精密度、稳定性、重复性良好。对不同产地和来源的草乌样品建立特征指纹图谱,并进行生物碱含量测定,有助于了解不同产地草乌之间的成分的相似性和差异,可以从一定程度上保证和控制一定区域内的草乌疗效和毒性。
