距离约束和二面角优化的蛋白质结构预测方法
2022-01-21周晓根张贵军
李 亭,刘 俊,周晓根,张贵军
1(浙江工业大学 信息工程学院,杭州 310023)2(密歇根大学 计算医学与生物信息学系,安娜堡 48109)
1 引 言
蛋白质是生命活动的主要承担者[1].蛋白质的三维结构则决定了其所具有的特定功能[2].获取蛋白质的三维结构是研究其生物机理的基础,对蛋白质功能注释、疾病研究、药物设计等方面有着重要的意义[3,4].目前主要通过生物实验测定方法获取蛋白质的三维结构,包括X-射线晶体衍射、核磁共振和冷冻电镜技术.这些实验测定方法耗时长且成本高昂[5-6].同时随着基因测序技术的快速发展,已测定蛋白质序列数目和已测定蛋白质结构数目之间存在巨大差距,并且这种差距正在逐年扩大.在理论探索和应用需求的推动下,根据Anfinsen法则[7],从氨基酸序列出发,利用计算机技术结合优化算法的从头蛋白质三维结构预测成为了生物信息学领域的研究热点[8].
从头蛋白质结构预测主要有两大难点:1)构建合适的能量函数,引导构象向天然态结构(即能量最低点状态)折叠;2)设计高效的构象搜索方法,在复杂的能量景观中探索低能量区域[9].传统的能量函数[10,11]是基于理化模型构建的,没有考虑到不同蛋白质的特殊性,因此很难准确地引导构象向天然态结构折叠.近几年,得益于已测定蛋白质序列的快速增长和深度学习技术的不断完善,根据目标序列的进化信息提取先验知识辅助结构预测取得了巨大进展[12-17].尤其是残基-残基接触预测,为蛋白质折叠提供了重要的约束信息,很大程度上弥补了能量函数不精确造成的影响.许多利用残基-残基接触预测蛋白质结构的方法被提出[18-21],其中CONFOLD[22]将残基-残基接触和二级结构信息转化为距离、二面角和氢键约束,进行蛋白质结构预测.自CASP13以来,基于深度学习的残基间距离预测迅速成为新的研究热点[23],残基间距离预测描述了残基对在不同距离区间的概率,相比残基-残基接触包含了更多的几何约束信息,更有利于结构预测[24].AlphaFold[25]根据预测的残基间距离分布构建蛋白质特定的势能函数,并利用随机梯度下降寻找最优结构,最终,AlphaFold在CASP13中脱颖而出.数十年来,研究学者们提出了大量方法进行蛋白质构象空间搜索,其中蒙特卡洛[26-28]和副本交换蒙特卡洛[29]取得了巨大成功,并成功的应用于国际知名的从头预测服务器Rosetta[30,31]和QUARK[32,33],而进化算法[34-40]、多模态优化[41]、分子动力学[42-44]模拟也展现出各自的优势.在差分进化算法框架下,SCDE[45]利用接触图和二级结构提出策略来预测蛋白质结构;ItFix[46]建立粗粒度模型,给定适当的搜索策略和能量函数进行结构预测.片段组装[47-49]策略被广泛应用于从头蛋白质结构预测方法,能够有效地减小构象搜索空间,提高采样效率.
针对上述问题,本文提出了一种基于距离约束和二面角优化的蛋白质结构预测方法(DCDA).首先,对预测的残基对距离分布信息进行筛选,构建基于残基对距离分布的构象评估模型,结合片段组装技术大范围搜索构象空间;进而,构建Loop区域特定的局部构象评估模型,并结合基于二面角的差分进化采样策略,增强结构灵活的Loop区域采样,突破片段库的约束,进一步提高预测模型的精度.15个测试蛋白质的实验结果表明,DCDA在预测精度上优于Rosetta、QUARK和CONFOLD,是一种有效的蛋白质结构预测方法.
2 DCDA算法设计
2.1 残基对距离特征提取
预测的残基对距离分布相比预测的残基对接触包含了更详细的几何约束信息,可以有效地减少构象搜索空间,更准确、快速地引导蛋白质折叠.为了避免冗余信息的影响,本文根据距离预测概率峰值对距离矩阵进行过滤.首先,去除掉残基对序列分离小于6的残基对;然后,对于预测的距离分布中任意残基对之间距离小于等于2的两个残基对,仅保留预测的距离概率峰值最大的残基对,两个残基对之间的距离计算公式如下:
(1)
其中,(i1,j1)表示两个残基对中的第1个残基对;(i2,j2)表示两个残基对中的第2个残基对.
图1为蛋白质1E2A_A过滤前后的残基对距离分布图.
2.2 基于残基对距离分布的构象评估模型
预测的残基对距离分布包含的是离散信息,即残基对距离分别在[2,2.5)Å、[2.5,3)Å、…、[19.5,20)Å区间内的概率.然而,结构预测过程中构象残基间的真实距离是连续的,为了更精确的评估构象,本文将离散的距离分布信息转换成连续的分布函数,确保对于每一个给定的距离都有与之对应的概率值.
本文采用三次样条插值为每一个残基对构造连续的距离分布函数.令每个预测距离区间的中值为三次样条插值的节点,分别在两端添加一个边界点,即节点分别为x0=0,x1=2.25,x2=2.75,…,x36=19.75,x37=∞;以38个节点为依据,构造37个三次函数fk(x):
fk(x)=ak+bkx+ckx2+dkx3,k={0,1,2,…,35,36}
(2)
其中,ak,bk,ck,dk为待定系数,共有148个待定系数.则残基对的距离分布函数可以表示为:
(3)
(4)
为了确定待定系数值,需构造148个方程进行求解.
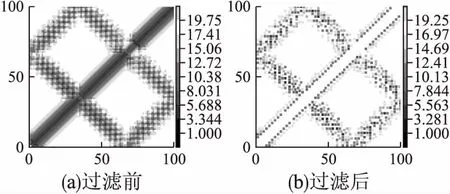
图1 过滤前后的残基对距离分布图Fig.1 Comparison distance map before and after filtering
首先,要求每一个分段函数经过对应的节点:
(5)
其中,pk表示节点xk对应的距离区间的概率值,令p0=0、p37=0;由公式(5)可构造74个方程.
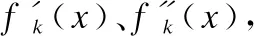
(6)
可构造72个方程.最后,通过边界条件再构造2个方程.通过求解148个方程得到系数值,得到方程表达式.
定义残基对(i,j)对应的距离分布函数为f(i,j)(x),x表示残基对(i,j)在构象中的真实距离.根据三次样条得到的方程来计算采样过程中构象的势能,构建构象评估模型,以引导构象向天然态折叠.
根据所有残基对的距离分布函数,构建构象评估模型:
(7)
其中Q是过滤后的残基对集合,dij为待评估构象的第i号残基和第j号残基的Cβ原子(甘氨酸为Cα原子)间的欧氏距离,f(i,j)(dij)是x=dij时f(i,j)(x)的函数值,表示残基对(i,j)的距离为dij的概率.
2.3 片段组装
由于蛋白质构象空间的高维特性,在巨大的构象空间中进行采样是不合适的.片段组装技术利用已知结构的局部信息,将每一个残基的二面角约束在一组离散值内,从而极大地缩小了构象搜索空间.对于给定目标构象,片段组装过程如下:首先,随机选择一个滑动窗口,在该窗口对应的片段库中随机选择一个片段替换窗口内原有的片段,如图2所示,L为序列长度;然后,利用公式(7)计算目标构象和组装得到的构象的得分,并根据boltzmann准则判断是否组装成功;如果成功,则用组装后的构象替换目标构象;否则,再次随机选择窗口进行片段组装,直至组装成功或组装次数达到200次.
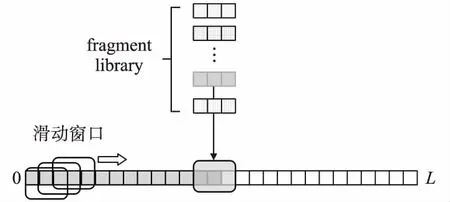
图2 片段组装示意图Fig.2 Schematic diagram of fragment assembly
2.4 基于二面角的差分进化采样策略
本文提出了基于差分进化算法的Loop区域二面角采样策略,在利用片段库信息的同时,避免结构灵活的Loop区域受到片段库的约束,探索更多结构合理的构象.首先,利用DSSP算法[50]计算目标构象的二级结构,随机选择一段二级结构为Loop的局部区域,并构建Loop区域特定的构象评估模型;然后,以窗口宽度为3个残基,步长为1个残基,从选定Loop区域的起始残基位滑动到结束残基位,形成K个滑动窗口.对每个窗口内的片段执行变异、交叉操作生成候选构象,添加到候选构象池;最终,通过选择操作,从候选构象池中选择最优构象替换目标构象.具体过程如下:
a)构建局部构象评估模型.针对选定的Loop区域,从距离分布图中选取残基编号分别位于选定区域左右两端的残基对构建局部评估模型.计算公式如下:
(8)
其中,M是选定区域左右两端的残基对集合,wij表示残基对(i,j)的权重,由残基i和残基j所在二级结构与选定Loop区域的相对位置关系决定的.计算公式如下:
(9)
其中,indexi表示残基对中残基i所在的二级结构片段与选定Loop结构之间非Loop结构的数量;indexj表示残基对中残基j所在的二级结构片段与选定Loop结构之间非Loop结构的数量.
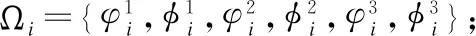
(10)
其中,F是缩放因子,一般在[0,2]之间选择,本文取0.5.
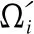
(11)
其中,CR表示交叉概率,l表示向量中角度的索引值,lrand表示一个随机整数,它的取值范围是[1,6].

d)选择操作.根据公式(8)计算目标构象和候选构象池中每个构象的得分,按得分由低至高选取候选构象池中的构象替换目标构象,直至替换成功或遍历完候选构象池中所有构象.候选构象替换目标构象的公式如下:
(12)
(13)
其中,KT表示温度常数.
2.5 算法描述
DCDA算法的流程描述如下:
输入:目标蛋白序列,片段库,残基预测距离
输出:蛋白质三维结构
1. 初始化种群T←Rosetta stage1,stage2
2.forg←1 to G do
3.fori←1 toNP
4.Ti←片段组装
5. 随机选取一段Loop区域,构建局部构象评估模型
6. 创建滑动窗口{W1,W2,…,WK}
7.fork←1 toK
11.endfor
13.fork←1 to K
16.break;
17.endif
18.endfor
19.endfor
20.endfor
21.return种群中得分最低的构象
3 实验结果与分析
3.1 测试蛋白和实验设置
为了验证算法的有效性,从PDB库中选取序列长度范围从72到147的15个蛋白质测试DCDA的性能.这15个测试蛋白质的折叠类型包括α、β和α/β,详细信息如表1中的第2-4列所示.这些蛋白质的三维结构已经由生物实验方法测定,且被广泛用于蛋白质测试[51,52],可以较好地测试DCDA的性能.
表1 测试蛋白的TM-score
Table 1 TM-score of test protein

No.PDBLenTypeDCDARosQUACONF11G8Q_A90α0.620.30.360.6221H4L_D147α0.590.270.420.6531W53_A84α0.70.420.370.6642BL7_A79α0.840.460.60.7951B4B_A72α/β0.820.330.410.6261CF7_B82α/β0.750.440.510.6571CQA_A123α/β0.680.270.350.5581DJ7_A109α/β0.550.270.380.3791IPI_A114α/β0.730.310.550.51101MWP_A96α/β0.510.260.340.3111PZW_A80α/β0.570.290.370.45122H8E_A120α/β0.660.30.340.57133CHB_D103α/β0.330.290.30.32141OK0_A74β0.630.260.220.22152BT9_A90β0.720.340.410.53Average0.6470.3210.3950.521
测试蛋白质的片段库和残基间距离分布信息分别从ROBETTA[53]和trRosetta[54]服务器获取,并且在构建片段库和距离分布信息的过程中均已去除同源模板.实验参数设置如下:种群规模NP=100、种群更新次数G=1000、温度常数KT=2、交叉概率CR=0.5、缩放因子F=0.5.在种群初始化过程中采用Rosetta score0和score1能量函数引导构象搜索.在片段组装和基于二面角的差分进化过程中分别采用构象评估模型Sd和局部评估模型Sloop_d引导构象采样.通过计算预测模型和天然态结构的Cα原子的均方根偏差(RMSD)和TM-score[55]来评估算法的预测精度.
3.2 实验结果分析
在测试蛋白质上,将DCDA算法的预测结果与Rosetta、QUARK和CONFOLD预测的结果进行了比较.Rosetta、QUARK和CONFOLD是蛋白质结构预测领域内认可度最高的方法之一,与其进行比较能更客观的反映DCDA的性能.Rosetta和QUARK均采用了片段组装技术,是经典的两个蛋白质结构预测服务器;CONFOLD则利用了残基接触、二级结构等先验信息构建几何约束,代表着新兴的预测算法.
表1和表2分别列出了DCDA、Rosetta、QUARK和CONFOLD在15个测试蛋白质上预测模型的TM-score和RMSD,其中Ros、QUA和CONF分别代表Rosetta、QUARK和CONFOLD.其中Rosetta的预测结果是通过安装本地版并使用默认参数预测得到的,QUARK和CONFOLD的预测结果是直接通过其在线服务器预测直接得到的.表中加粗的数据表明在对应指标下,相应算法的预测精度最优.
如表1所示,DCDA在15个测试蛋白质中的14个上获得了TM-score最高的预测模型,尤其对于2BL7_A和1B4B_A,DCDA预测模型的TM-score均在0.8以上;DCDA预测出了14个测试蛋白质的正确折叠(TM-score≥0.5),占全部测试蛋白的93.3%,是所有对比方法中最高的;在平均TM-score上,相比于其余3种方法中预测精度最高的CONFOLD提高了24.2%.由此可见,DCDA预测模型的TM-score明显优于Rosetta、QUARK和CONFOLD.表2列出了4种方法预测模型与天然态结构的RMSD.DCDA在14个测试蛋白质上预测出了RMSD最小的结构模型,平均RMSD为4.288Å,相比较于CONFOLD的平均RMSD降低了46.1%.尤其是蛋白1OK0_A,DCDA的RMSD相比QUARK降低了7.01Å.由此可见,在RMSD方面,DCDA算法的预测精度优于Rosetta、QUARK和CONFOLD算法.整体而言,DCDA在TM-score和RMSD评价指标上均优于对比算法,是一种有效的蛋白质结构预测算法.
表2 测试蛋白的RMSD
Table 2 RMSD of test protein

No.PDBDCDARosQUACONF11G8Q_A4.0611.4310.654.521H4L_D5.215.9210.894.8731W53_A3.2511.1312.075.3642BL7_A2.037.53.372.4651B4B_A2.859.328.163.9861CF7_B2.797.386.363.2171CQA_A4.2613.558.026.0881DJ7_A6.3413.4210.6612.2691IPI_A3.3611.834.8514.52101MWP_A6.1513.5414.8813.57111PZW_A5.3911.137.519.03122H8E_A3.7814.3510.348.87133CHB_D8.9713.3512.3214.11141OK0_A3.3510.7210.3611.56152BT9_A2.5410.126.824.96Average4.28811.6469.1517.956
为了验证基于二面角的差分进化采样策略的有效性,本文设计了未使用基于二面角的差分进化采样策略的对比实验DC,对比结果如表3所示.与DC算法相比,DCDA在15个测试蛋白质中的11个蛋白质上获得了RMSD最小的结构模型,在13个蛋白质上获得了TM-score最高的预测模型.对比表2和表3的结果可以看出,仅使用残基间距离分布信息的DC算法,在15个测试蛋白质中,14个测试蛋白质的TM-score比Rosetta高,其平均TM-score值比Rosetta高0.275,其平均RMSD比Rosetta低6.267Å.
表3 DCDA组件比较
Table 3 Comparison of the DCDA component
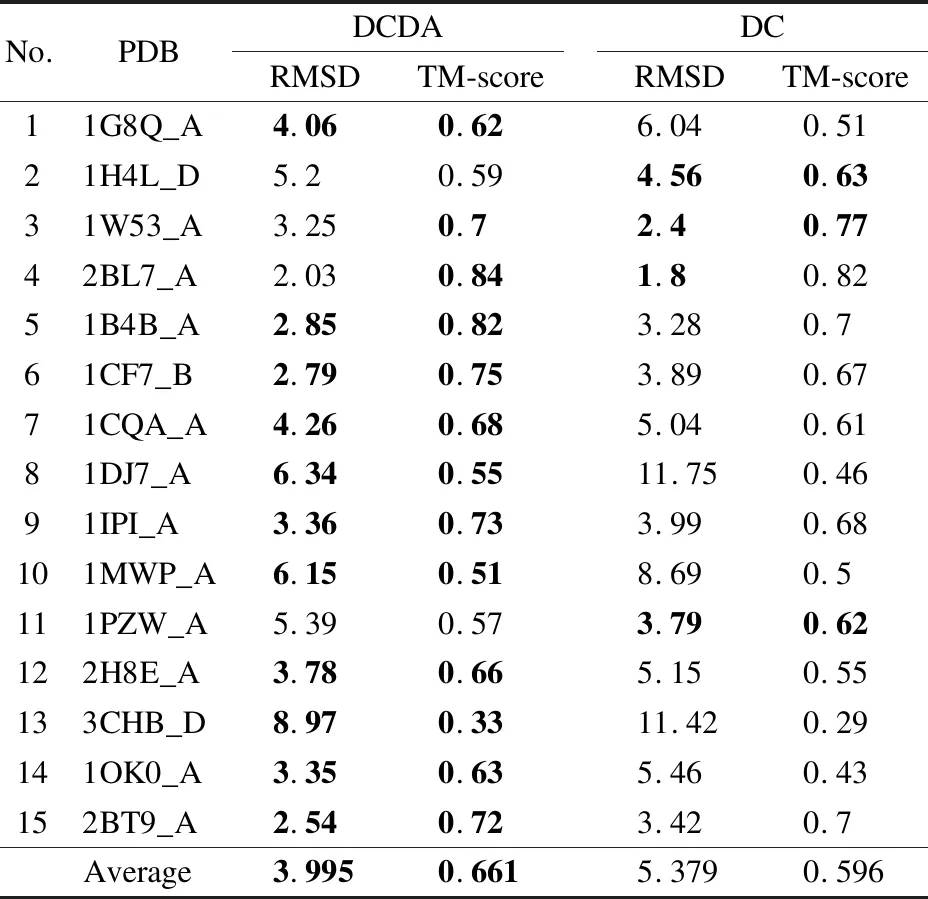
No.PDBDCDARMSDTM-scoreDCRMSDTM-score11G8Q_A4.060.626.040.5121H4L_D5.20.594.560.6331W53_A3.250.72.40.7742BL7_A2.030.841.80.8251B4B_A2.850.823.280.761CF7_B2.790.753.890.6771CQA_A4.260.685.040.6181DJ7_A6.340.5511.750.4691IPI_A3.360.733.990.68101MWP_A6.150.518.690.5111PZW_A5.390.573.790.62122H8E_A3.780.665.150.55133CHB_D8.970.3311.420.29141OK0_A3.350.635.460.43152BT9_A2.540.723.420.7Average3.9950.6615.3790.596
实验数据表明基于二面角的差分进化采样策略可以有效探索片段库约束空间之外的构象,提高结构灵活的Loop区域结构的多样性,进而提高预测模型的精度.此外,对比表3中DC与表1和表2中Rosetta、QUARK、CONFOLD的结果可以发现,仅使用残基间距离分布信息的方法预测的结果也明显优于对比的方法,进一步表明预测的残基间距离分布信息能够更好的引导构象采样;并且,结合基于二面角的差分进化采样策略能够进一步提升预测精度.
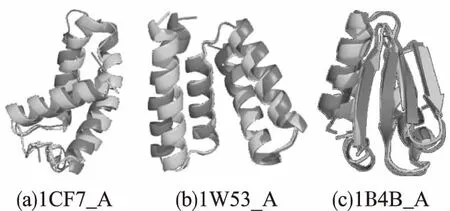
图3 DCDA预测结构与天然态结构的比对Fig.3 Comparison between predicted structure by DCDA and native structure
图3是DCDA预测的模型与天然态结构之间的比对图,其中包含α和α/β折叠类型.从图中可以发现DCDA预测的蛋白质结构模型与天然态结构之间有着较好的重叠,和天然态结构相似,具有很好的匹配度.
3.3 算法采样能力分析
本节对DCDA和Rosetta的采样能力进行了分析和对比.部分蛋白的采样分布如图4所示,图中横坐标为搜索过程中产生的构象与天然态结构之间的RMSD,纵坐标为每个区域内构象数占所有构象总数的百分比,实线是DCDA的采样分布,虚线是Rosetta的采样分布.可以发现,DCDA在4个测试蛋白上的近天然态采样比例均高于Rosetta,DCDA采样概率峰值的RMSD比Rosetta更接近零,这表明DCDA的近天然态采样能力强于Rosetta.对于蛋白质1W53_A和2BL7_A,DCDA分别有89.95%和91.3%的过程构象的RMSD小于5Å,而Rosetta在1W53_A上的过程构象的RMSD均大于5Å,在2BL7_A上的过程构象的RMSD仅有5.51%小于5Å,对于1CQA_A,尽管DCDA和Rosetta的近天然态采样能力都不强,但是DCDA的采样分布比Rosetta更接近天然态构象.

图4 RMSD分布比较Fig.4 Comparison of RMSD distributions
总体而言,DCDA在搜索过程中能够采样到更多近天然态构象.主要是DCDA算法采用了距离分布信息和基于二面角的差分进化采样策略,有效的提升了预测精度.
4 结 论
本文提出一种距离约束和二面角优化的蛋白质结构预测方法.首先,对预测的距离分布图进行筛选;然后,利用三次样条插值根据预测的蛋白质残基间距离分布构建构象评估模型,结合片段组装技术,大规模采样构象空间;进而,在进化算法的框架下,针对结构灵活的Loop区域,设计基于二面角的差分进化采样策略,构建局部构象评估模型,利用基于滑动窗口的二面角交叉变异生成多个候选构象,并根据局部构象评估模型实现构象更新.基于二面角的差分进化采样策略能够突破片段库的约束,采样到更多近天然态构象,提高算法的预测精度.在15个测试蛋白质上的实验结果表明,DCDA算法具有较强的搜索性能和较高的预测精度,是一种有效的构象空间搜索算法.在下一步研究中,我们将利用深度学习技术构建深度残差神经网络预测蛋白质残基二面角,根据预测的二面角和距离信息构建约束函数,进一步提高蛋白质结构预测精度.
