测定食品中16种氨基酸国家标准方法的改进
2022-01-19李巧琪李洪燕卢桂锋周海练许志彬
李巧琪,李洪燕,卢桂锋,周海练,许志彬
(广州检验检测认证集团有限公司 国家加工食品质量检验中心(广东),广州 511447)
自然界存在300多种氨基酸,其中,构成人体蛋白质的有20多种,被称为基本氨基酸[1]。从营养学角度看,由于部分氨基酸人体无法直接合成,需要从食品中摄取,并且食品中蛋白质的营养价值取决于食品中所含氨基酸的种类和含量,因此测定食品中多种氨基酸的含量对人体营养摄入评估具有一定意义[2]。
目前,食品中氨基酸的前处理方法主要采用酸水解法[3],但是该方法前处理时间较长(需要22 h左右)。测定方法主要有液相色谱-质谱法[4-6]、液相色谱法[7-9]、离子色谱法[10-12]等。其中液相色谱-质谱法仪器成本较高;而液相色谱法中,样品经柱前衍生反应后,部分产物受基体以及过量试剂的影响,稳定性较差,并且衍生试剂的价格较高;阴离子交换直接检测法易受细菌干扰,进而造成测定结果重现性不理想。基于此,本工作首先对国家标准GB 5009.124-2016《食品安全国家标准 食品中氨基酸的测定》中的酸水解法进行改进,并提出了微波消解这一前处理方法,然后采用优化后的阳离子交换色谱分离-柱后茚三酮衍生法测定食品中天冬氨酸(Asp)、苏氨酸(Thr)、丝氨酸(Ser)、谷氨 酸(Glu)、脯氨酸(Pro)、甘氨酸(Gly)、丙氨酸(Ala)、缬氨酸(Val)、蛋氨酸(Met)、异亮氨酸(Ile)、亮氨酸(Leu)、酪氨酸(Tyr)、苯丙氨酸(Phe)、组氨酸(His)、赖氨酸(Lys)、精氨酸(Arg)等16种氨基酸的含量。该方法分析结果稳定,重现性好,为食品中氨基酸的快速测定提供了参考。
1 试验部分
1.1 仪器与试剂
S433D 型氨基酸分析仪;ED53 型精密烘箱;TVE-1100型试管浓缩仪;Ethos MHP型微波蛋白质水解系统;ME204E型电子天平。
17种氨基酸的混合标准溶液:胱氨酸(Cys)的浓度为1.25 mmol·L-1,Asp、Thr、Ser、Glu、Pro、Gly、Ala、Val、Met、Ile、Leu、Tyr、Phe、His、Lys、Arg的浓度均为2.50 mmol·L-1。
缓冲液1:称取柠檬酸三钠11.8 g、柠檬酸6.0 g于烧杯中,加入65 mL乙醇、5.6 mL盐酸以及适量水,溶解后用水定容至1 L,用6 mol·L-1盐酸溶液将溶液酸度调至pH 3.42。
缓冲液2:称取柠檬酸三钠19.6 g、硼酸5.0 g、氢氧化钠3.1 g于烧杯中,用水溶解并定容至1 L,用20%(质量分数)氢氧化钠溶液将溶液酸度调至pH 10.85。
再生液:称取氢氧化钠20.0 g于烧杯中,用水溶解并定容至1 L。
显色液:称取乙酸钾196 g、三水合乙酸钠272 g于烧杯中,加入乙酸200 mL,用水溶解并定容至1 L,配制成钾钠缓冲液,备用;称取茚三酮20 g、苯酚2 g于另一烧杯中,加入甲醇600 mL和钾钠缓冲液400 mL,溶解,配制成显色液,避光保存。
氮气的纯度不小于99.999%;甲醇为色谱纯;盐酸、柠檬酸三钠、氢氧化钠、硼酸、乙醇、柠檬酸、乙酸钾、三水合乙酸钠、乙酸、苯酚、茚三酮均为分析纯;试验用水为超纯水。
1.2 仪器工作条件
LCA K06/Na 色谱柱(150 mm×4.6 mm,5μm);氮气增压60~80 kPa。柱升温程序:初始温度58 ℃,保持22.0 min;22.0~27.0 min,升温至73 ℃,保持17.0 min;44.0~48.0 min,降温至58 ℃。流动相A为缓冲液1,B为缓冲液2,C 为 再生液,流量0.45 mL·min-1,压力3~4 MPa;衍生体系为显色液,流量0.25 mL·min-1,压力0.7~1 MPa;检测波长为440 nm(Pro)和570 nm(其余15种氨基酸);进样量50μL。梯度洗脱程序见表1。
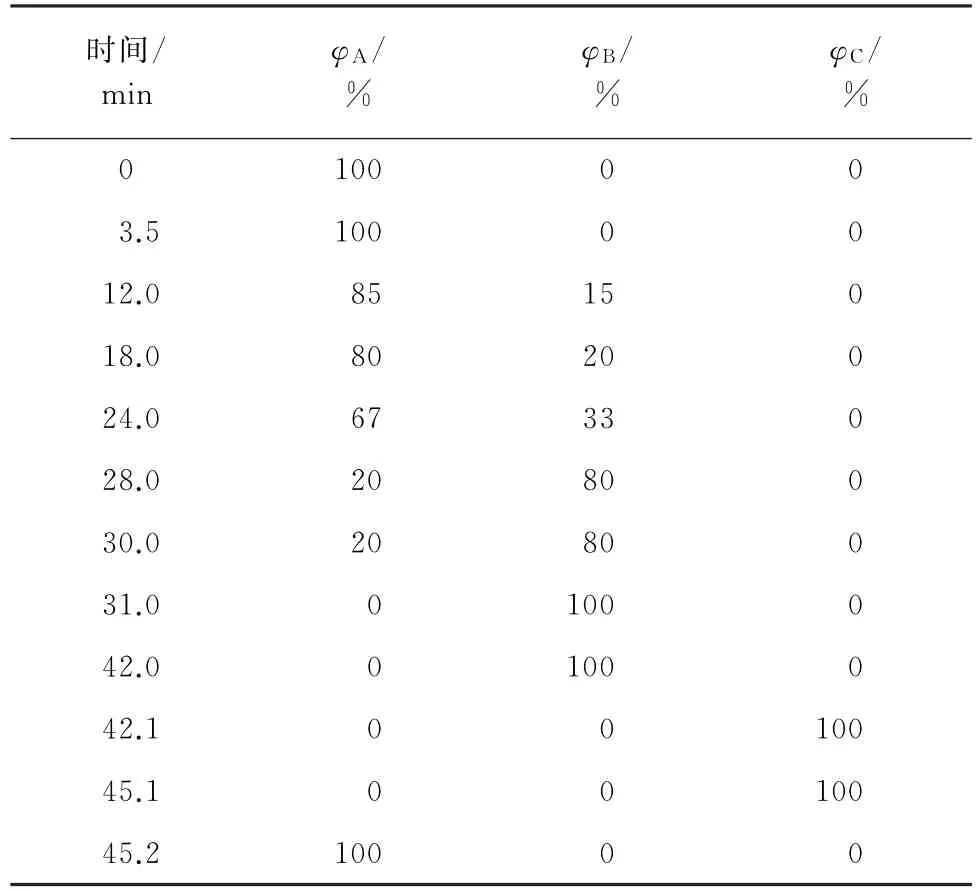
表1 梯度洗脱程序Tab.1 Gradient elution program
1.3 试验方法
1.3.1 常规酸水解法
参照GB 5009.124-2016中的方法进行测定。
1.3.2 改进后的酸水解法
称取样品1 g于聚四氟乙烯管中,加入含0.1%(质量分数)苯酚的6 mol·L-1盐酸溶液10 mL,充氮后封口,于165℃水解1 h。水解结束后,取出,冷却,用水定容至50 mL,取1 mL 减压浓缩至近干,再用10 mL 0.02 mol·L-1盐酸溶液复溶,过滤,滤液按照仪器工作条件进行测定。根据样品的实际情况可进一步稀释。
1.3.3 微波消解法
称取样品约0.1 g于石英罐中,加入6 mol·L-1盐酸溶液1 mL,充分湿润后,将其放入装有6 mol·L-1盐酸溶液的聚四氟乙烯罐中,充氮除氧,封盖后放入微波消解仪,设置微波输出功率为1 000 W,于165 ℃消解12 min。消解结束后,取出,冷却,将消解液转移至50 mL 容量瓶中,用0.02 mol·L-1盐酸溶液稀释至刻度,摇匀,过滤,滤液按照仪器工作条件进行测定。根据样品实际情况可进一步稀释。
2 结果与讨论
2.1 酸水解条件的优化
提高水解温度可使活化分子增多,水解效率提高。为防止普通安培管在高温下难以承受盐酸蒸气压而破裂,试验采用聚四氟乙烯管进行水解。以猪瘦肉为研究对象,考察了不同酸水解条件对样品中16种氨基酸测定结果的影响,并与GB 5009.124-2016的测定结果进行比对,结果见表2。
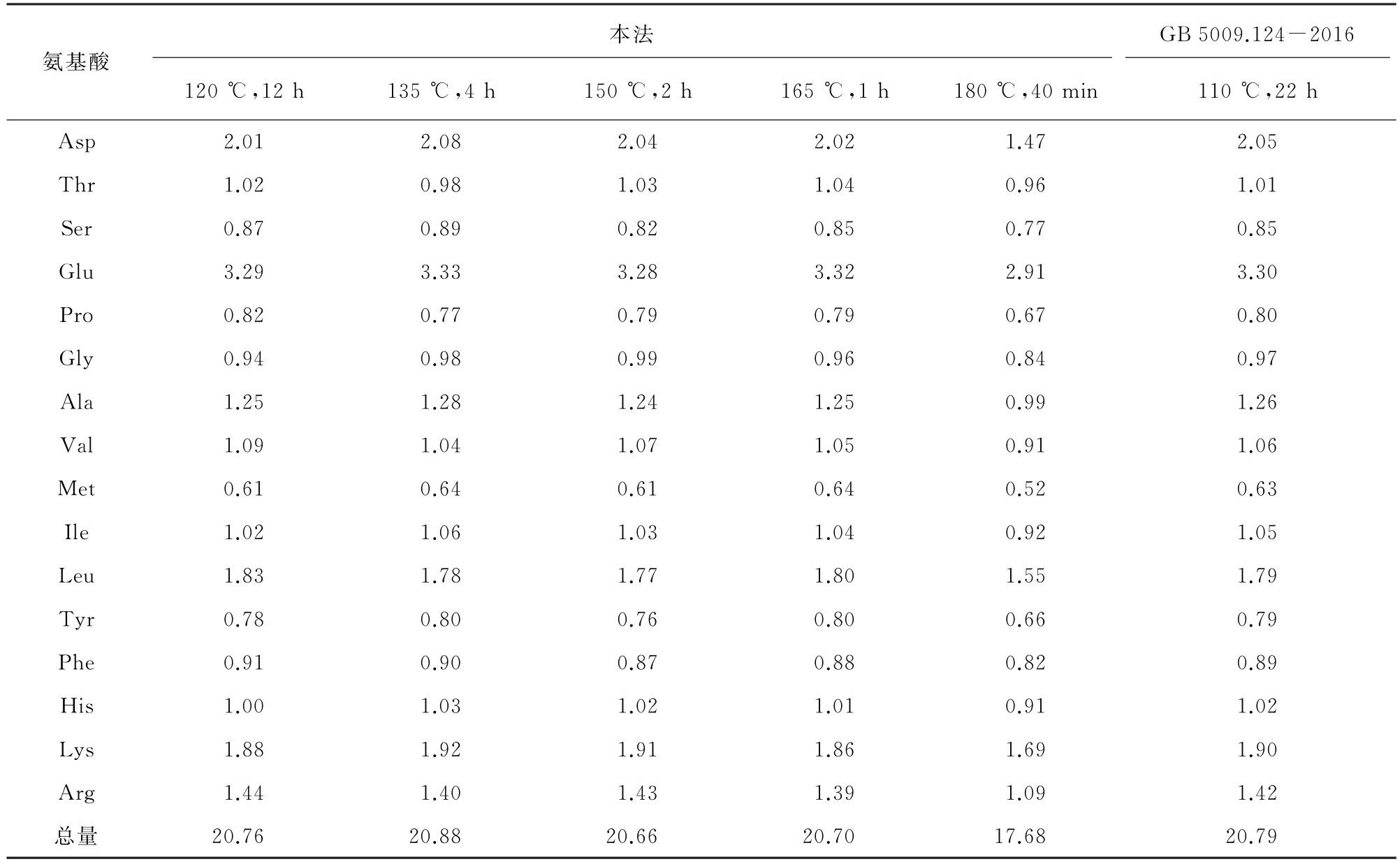
表2 不同酸水解条件下氨基酸的测定结果Tab.2 Determination results of amino acids under different acid hydrolysis conditions %
结果表明:随着酸水解温度的升高,水解时间逐渐缩短,酸水解效率逐渐提高;当酸水解温度为180 ℃时,部分氨基酸分解,导致其测定值有所下降。因此,试验选择酸水解温度为165℃,酸水解时间为1 h,此条件下与GB 5009.124-2016的测定结果基本一致,且酸水解时间由22 h缩短至1 h。
2.2 微波消解条件的优化
微波消解法是常见的样品前处理技术,其消解效率远远高于酸水解法。设置微波输出功率为1 000 W,以酸水解最佳温度165 ℃为消解温度,考察了不同消解时间(6,9,12,15,18 min)对2.1节同一猪瘦肉样品中16种氨基酸测定结果的影响,并与GB 5009.124-2016的测定结果进行比对,结果见表3。
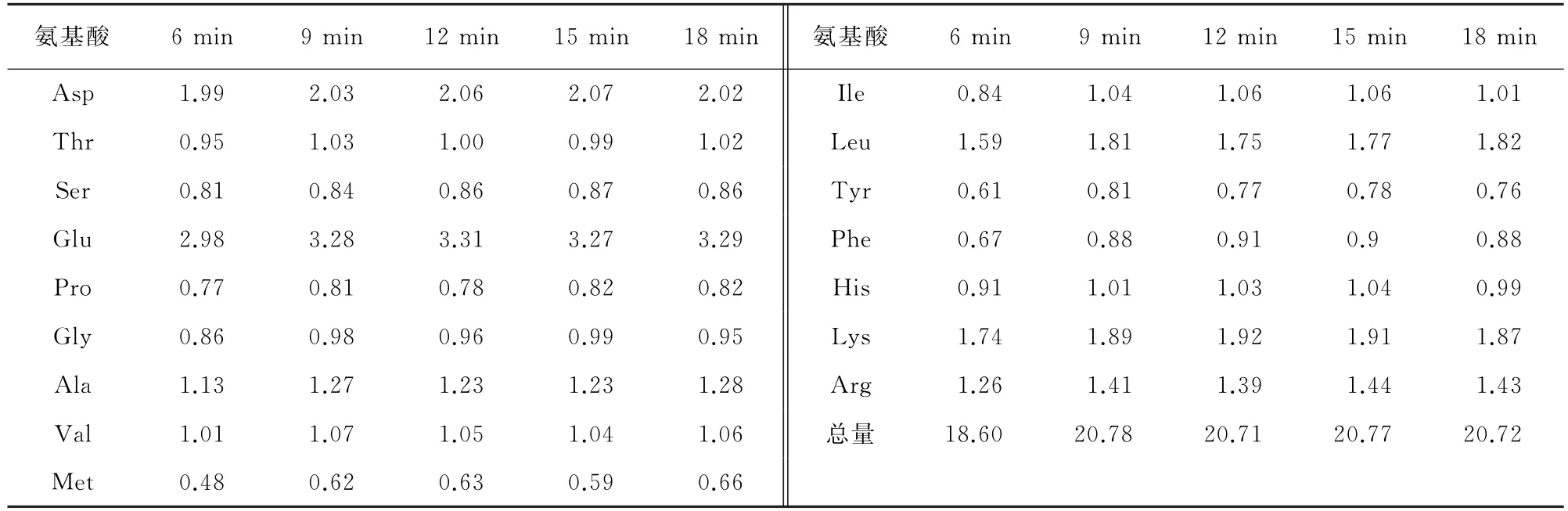
表3 不同消解时间下氨基酸的测定结果Tab.3 Determination results of amino acids under different digestion time %
由表3可知:当消解时间为6 min时,蛋白质消解不彻底,测定值比GB 5009.124-2016的测定值小;当消解时间为9~18 min 时,测定值与GB 5009.124-2016的测定值基本一致。为避免因消解时间过短而出现复杂基质样品消解不彻底现象,同时提高分析效率,试验选择消解时间为12 min。
2.3 柱温的优化
参考GB 5009.124-2016,采用阳离子交换色谱分离-柱后茚三酮衍生法进行分析。试验发现,柱温过低,色谱峰峰形变宽变矮。特别是Asp对温度十分敏感,当柱温不大于30 ℃时,其色谱峰在Thr后面出峰;当柱温不小于45 ℃时,其色谱峰在Thr前面出峰;当柱温达到58 ℃时,Asp和Thr分离更彻底。并且,随着柱温的继续升高,Thr和Ser出现叠峰现象;但当柱温控制在58 ℃时,Tyr和Phe出现叠峰现象,需要进一步升温才能使两者完全分离,经试验,当柱温达73℃,两者才可分离完全。因此,试验采用柱升温程序来分离各目标物,柱升温程序见1.1节。
2.4 洗脱程序的优化
无论是酸水解法还是微波消解法,都会将部分Cys转化为磺基丙氨酸,造成Cys的测定值偏小。并且试验过程中发现,Cys的保留时间与Val相近,流动相A 的酸度及流动相B 的更换时间对Cys的保留时间影响较大。为避免Cys对Val测定结果的影响,试验将Cys作为杂质,考察了流动相A 的酸度及流动相B 的更换时间对各目标物分离效果的影响。结果显示:当流动相A 的pH 小于3.30或流动相B的更换时间不小于14.0 min时,Cys的保留时间较长,并且与Val的色谱峰黏连在一起,造成Val的测定值偏大;当流动相A 的pH 大于3.50或流动相B的更换时间不大于10.0 min时,各目标物的保留时间均提前,但Thr与Ser、Leu与Ile分离不完全;当流动相A 的pH 为3.40~3.45以及流动相B的更换时间为12.0 min时,Cys与Val分离效果较好。因此,试验选择流动相A 的酸度为pH 3.42,流动相B的更换时间为12.0 min,梯度洗脱程序见表1。
在上述优化条件下对17种氨基酸的混合标准储备溶液进行测定,所得色谱图见图1。
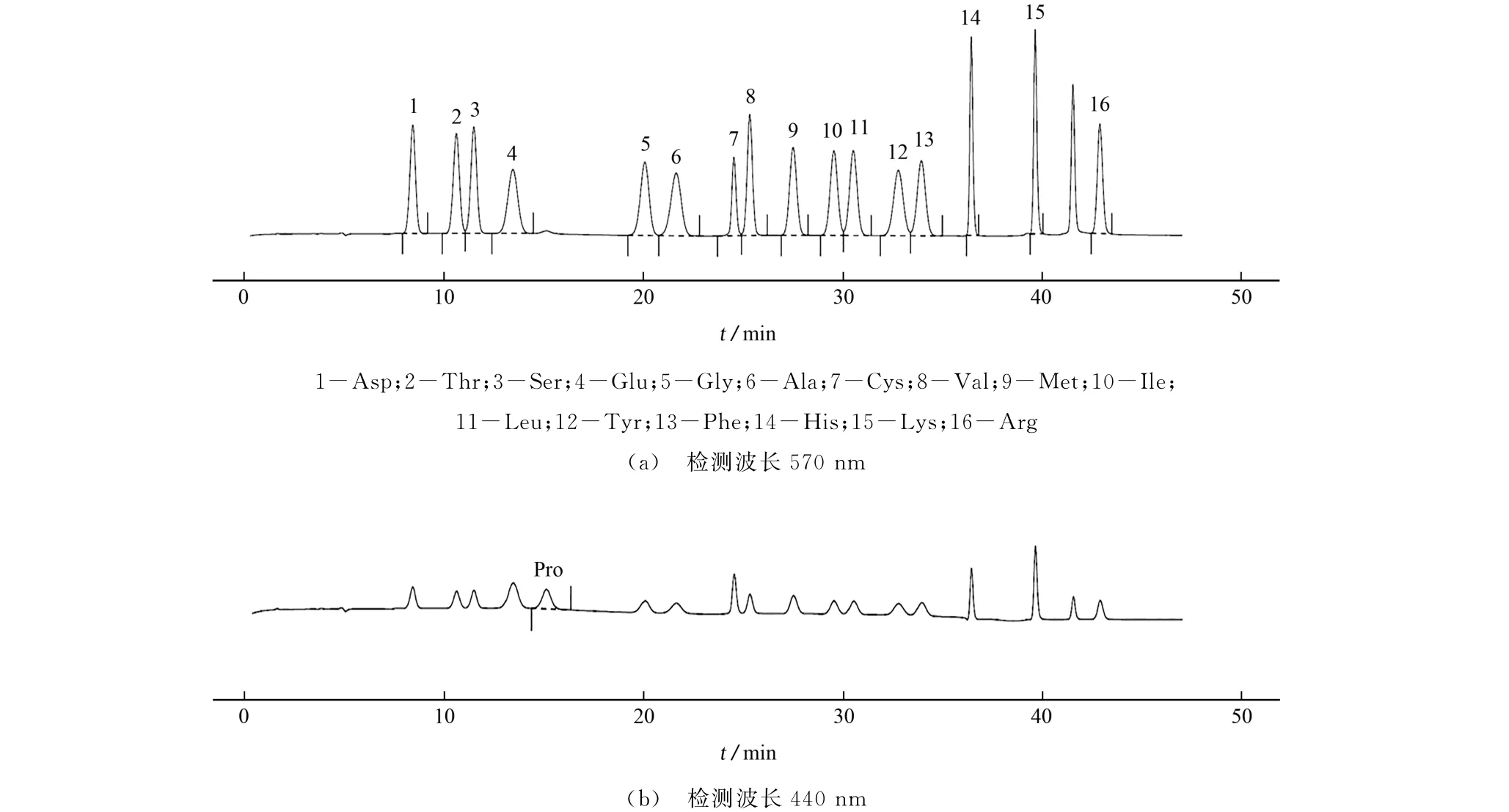
图1 色谱图Fig.1 Chromatograms
2.5 标准曲线与检出限
移取适量的17 种氨基酸的混合标准溶液,用0.02 mol·L-1盐酸溶液逐级稀释,配制成16种目标物浓度为10,20,50,100,200μmol·L-1的混合标准溶液系列。按照仪器工作条件对上述混合标准溶液系列进行测定,以各目标物的浓度为横坐标,其对应的峰面积为纵坐标绘制标准曲线。结果显示,16种氨基酸的浓度在10~200μmol·L-1内与其对应的峰面积呈线性关系,线性参数见表4。
通常情况下,食品中均含有氨基酸,无法采用阴性样品进行检出限试验。因此,本工作分别按照改进后的酸水解法和微波消解法对猪瘦肉进行处理,采用2倍基线噪声峰高(峰底附近30个数据点以内的值)乘以样品含量(浓度乘以进样量),再除以目标物峰高来计算检出限,并通过配对样本t检验法验证上述两种方法检出限与GB 5009.124-2016检出限的差异性,结果见表4。
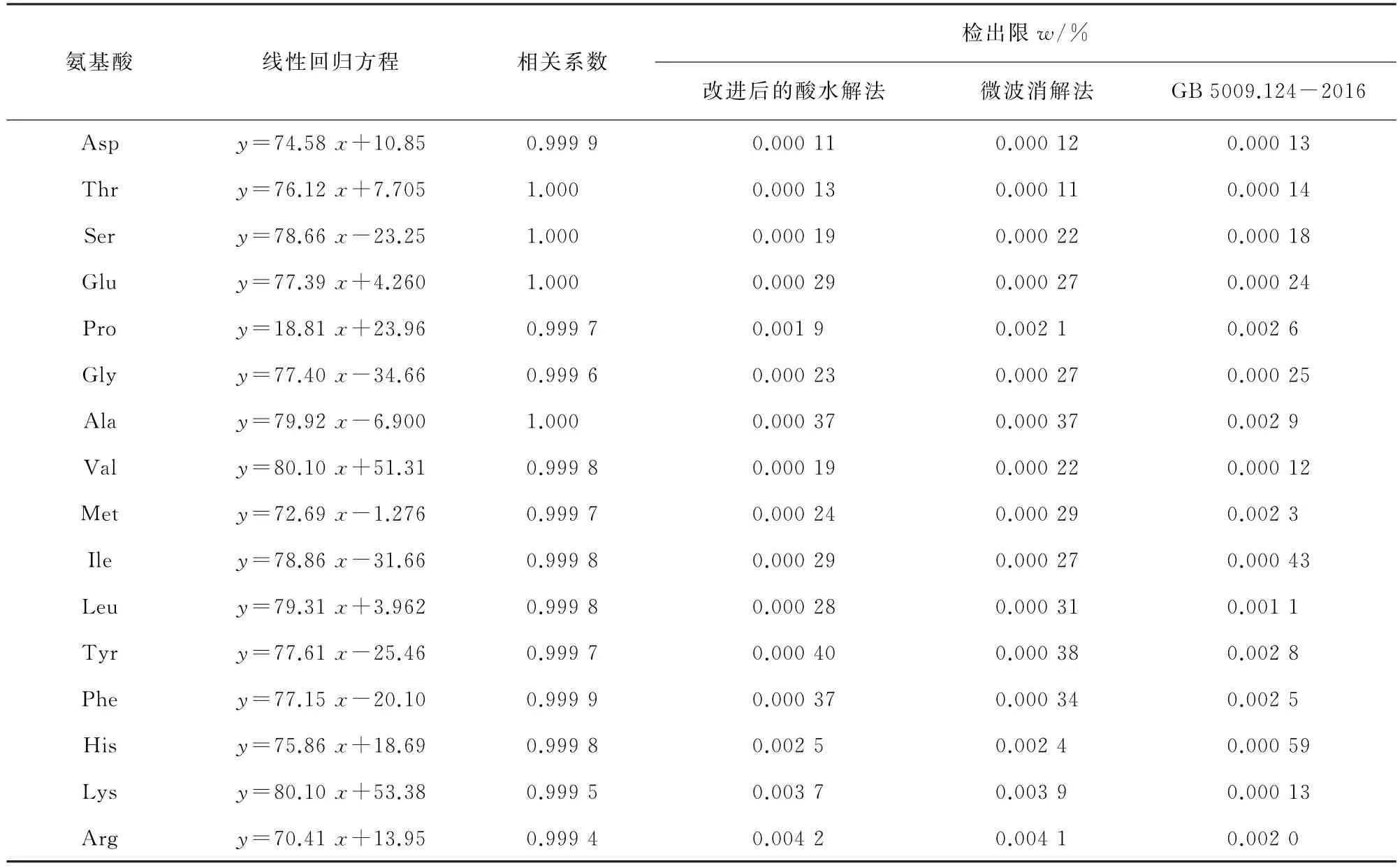
表4 线性参数和检出限Tab.4 Linearity parameters and detection limits
结果显示,改进后的酸水解法和微波消解法所得检出限与GB 5009.124-2016的检出限相当[3],P值依次为0.589,0.621,说明这两种方法与GB 5009.124-2016不存在显著性差异(P>0.05),能够满足日常检测需要。
2.6 精密度和回收试验
分别按照改进后的酸水解法和微波消解法对猪瘦肉进行低(20μmol·L-1)、中(50μmol·L-1)、高(100μmol·L-1)等3个浓度水平的加标回收试验,每个浓度水平平行测定6次,计算回收率和测定值的相对标准偏差(RSD),结果见表5。
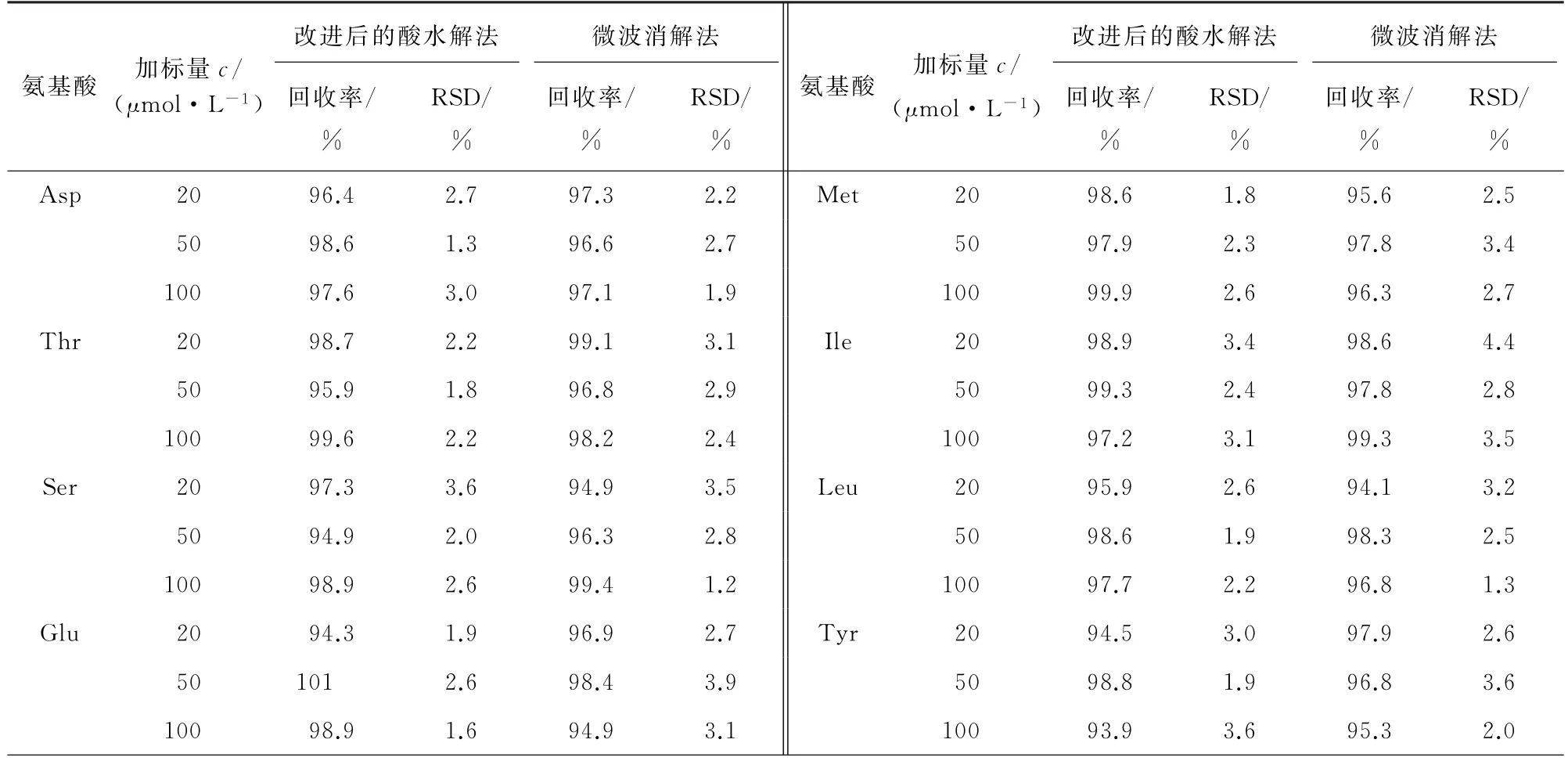
表5 精密度和回收试验结果(n=6)Tab.5 Results of tests for precision and recovery(n=6)
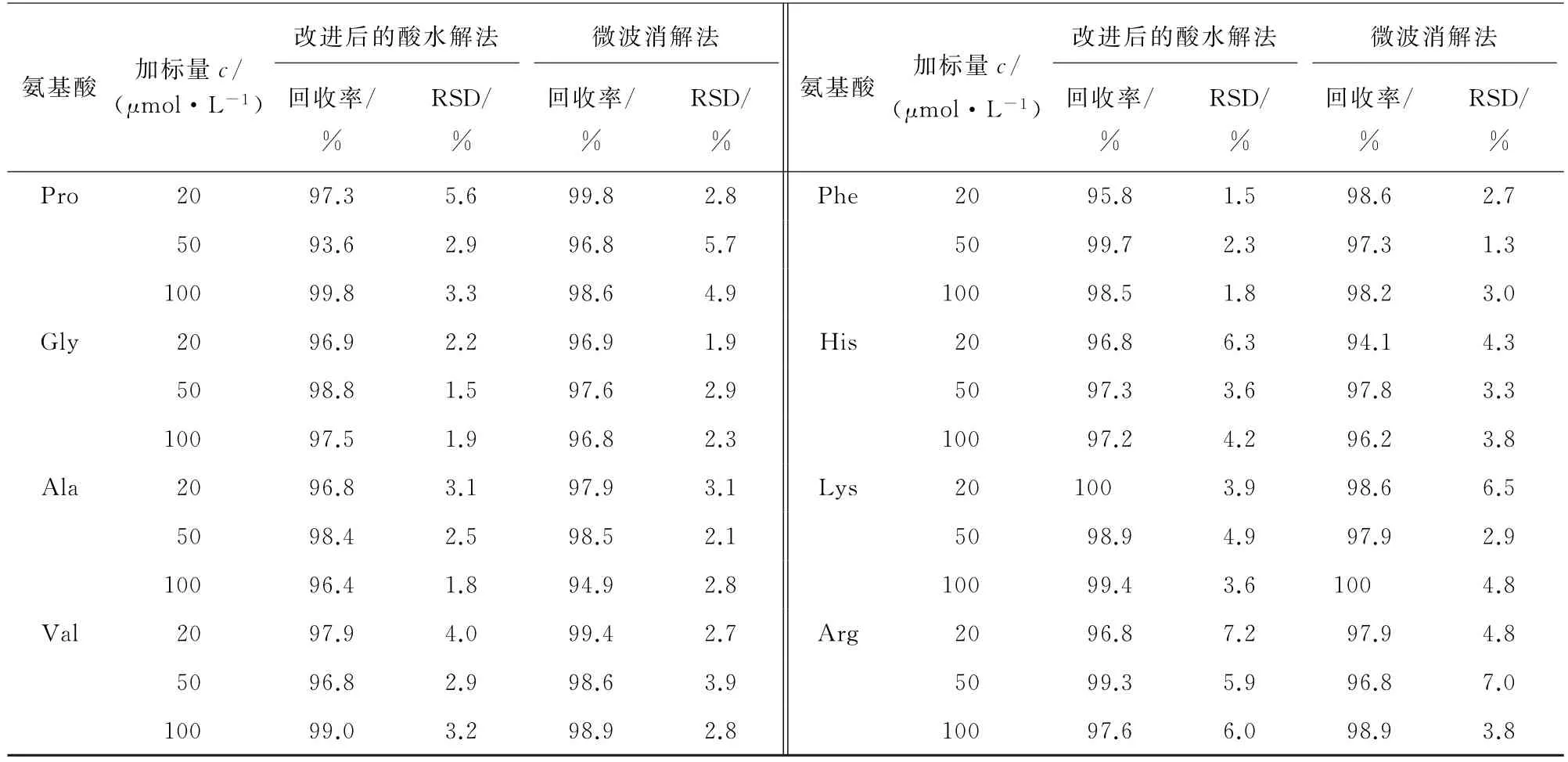
表5 (续)
结果显示:采用改进后的酸水解法时,16种氨基酸的回收率为93.6%~101%,测定值的RSD 为1.3%~7.2%;采用微波消解法时,16种氨基酸的回收率为94.1%~100%,测定值的RSD 为1.2%~7.0%,两种方法的准确度和精密度均符合分析检测要求[13]。
2.7 样品分析
按照改进后的酸水解法、微波消解法和GB 5009.124-2016,分别对不同食品中16种氨基酸进行测定,计算16种氨基酸总量,并通过配对样本t检验法分析改进后的酸水解法、微波消解法与GB 5009.124-2016是否存在显著性差异,结果见表6。
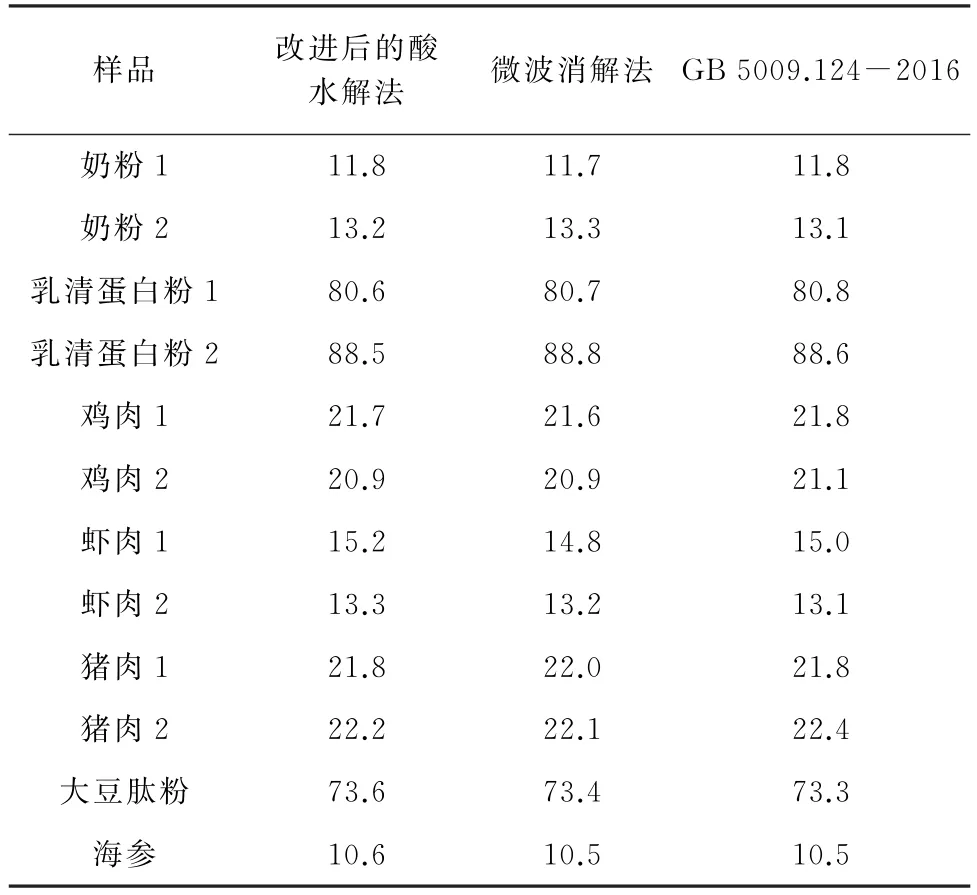
表6 样品分析结果Tab.6 Analytical results of samples %
结果显示,改进后的酸水解法、微波消解法与GB 5009.124-2016的测定结果基本一致,并且计算得改进后的酸水解法与GB 5009.124-2016的P值为0.870,微波消解法与GB 5009.124-2016的P值为0.643,均大于0.05,说明这两种方法的测定结果与GB 5009.124-2016的不存在显著性差异,进一步验证了这两种方法的准确度较好。
本工作通过对GB 5009.124-2016中的常规酸水解法进行改进,将原来的前处理时间由22 h缩短至1 h(改进后的酸水解法)或12 min(微波消解法),并进一步优化柱温和洗脱程序,建立了阳离子交换色谱法测定食品中16 种氨基酸含量的方法。改进后的酸水解法和微波消解法大大提高了试验效率,并且回收率高,精密度好,适用于食品中16种氨基酸的快速测定。
