广义布洛赫条件下二维晶格的磁交换作用
2022-01-19赵红艳蒋灵子朱岩潘燕飞樊济宇马春兰
赵红艳 蒋灵子 朱岩† 潘燕飞 樊济宇 马春兰
1)(南京航空航天大学理学院物理系,南京 210006)
2)(南京航空航天大学,空天信息材料与物理工信部重点实验室,南京 210006)
3)(苏州科技大学物理科学与技术学院,江苏省微纳米热流技术与能源应用重点实验室,苏州 215009)
二维磁性材料是近几年新兴的研究领域,该材料在开发自旋电子器件等领域具备良好的应用潜能.为了了解二维磁性材料的磁性质,明确体系内各近邻磁性原子间的磁相互作用非常重要.第一性原理为各近邻磁交换参数的计算奠定了基础.目前各近邻参数的第一性原理计算常用的是能量映射法,但这种方法存在一定的缺陷.本文通过广义布洛赫条件推导了3 种常见二维磁性结构的海森伯作用与Dzyaloshinskii-Moriya (DM)相互作用的自旋螺旋色散关系,这3 种结构为四方结构,元胞包含一个磁性原子的六角结构,元胞包含两个磁性原子的六角结构.为了将本文推导的自旋螺旋色散关系应用于实际,我们通过第一性原理计算了一些材料的海森伯和DM作用的交换参数,这些材料分别是MnB,VSe2,MnSTe,Cr2I3Cl3.其中,MnSTe 和Cr2I3Cl3 都属于二维Janus 材料,磁性原子层的上下层对称性破缺,整个体系存在DM 相互作用.
1 引言
二维磁性材料是指具有本征磁性的二维材料.由于维度的特殊性,二维磁性材料更容易受到其接触材料的影响,从而能够产生新奇的现象,例如非常规超导和反常霍尔效应等.因此,二维磁性材料为研究这些物理现象提供了良好的平台.同时,二维磁性材料的物理特性容易控制,例如,通过电场或者静电掺杂可以改变材料的交换参数和磁各向异性[1],或通过压力与应变改变材料的磁性.可操控的物理性质使得二维磁性材料更容易形成异质结.某些异质结不仅具有研究价值,更是在工程应用中具有良好的发展前景[2].部分二维磁性半导体材料的电子运输和磁性之间存在强耦合,这一性质有助于提升场效应管在低温和磁性材料中的性质[3].同时,一些二维磁性材料,如Fe3GeTe2材料,同源连接可用于制造多态垂直自旋阀,有助于将自旋信息拓展到二维材料[4].在开发一些自旋电子设备、磁控装置和微电子器件等领域,二维磁性材料都具有特殊意义.
早期发现的二维材料并不具有磁性,人们致力于通过外部诱导在二维材料中制造空穴来掺杂磁性原子,然而这种方法产生的磁性不具有长程有序性.根据Mermin-Wagner 定理[5],由于热激发,各向同性的二维材料不可能在有限温度下达到长程磁有序.因此,得到长程磁有序的二维磁体需要破坏各向同性的条件.近年来,相关的研究得到了重大的突破.2017年,美国加州大学的张翔院士课题组[6]通过磁光克尔技术发现了本征长程磁有序的Cr2Ge2Te6晶体,双层的Cr2Ge2Te6材料在低温下呈现顺磁性,其居里温度可通过外场调控,这一性质在自旋电子器件的研发中有望得到应用.在同一时期,华盛顿大学的徐晓东课题组[7]通过机器剥离法制备出了单层的CrI3晶体,CrI3晶格是A 型反铁磁体,但单层显示为铁磁性.2018年,张远波团队[8]通过离子栅压来调控剥离了二维磁性材料Fe3GeTe2,这种二维材料中的流动电子和原子的内在磁矩共存并相互作用,可能导致大尺度的相变和新的现象.Fe3GeTe2是金属铁磁体,改变晶体中Fe 原子的浓度可以调控材料的居里温度和矫顽力.同时,单层的VSe2[9],MnSe2[10]也成功被制备.Fe3GeTe2,VSe2和MnSe2的居里温度都可被调控到室温以上,这为相关的研究提供了更多的便利.
磁性原子之间相互作用的哈密顿量一般表示为
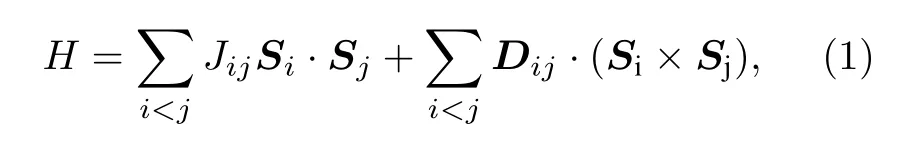
其中Si和Sj分别为晶格格点i和格点j上的自旋算符.(1)式中的第一项是各向同性的海森伯相互作用项(Heisenberg interaction,HBI).第二项是Dzyaloshinskii-Moriya 相互作用项(DMI).
Dzyaloshinskii-Moriya 相互作用又称手性磁相互作用,是在海森伯模型的基础上考虑自旋轨道耦合条件而产生的相互作用项[11,12].在不考虑自旋轨道耦合的情况下,电子在跃迁时其自旋取向保持不变,因此电子只能在自旋取向相同的轨道上跃迁.在自旋轨道耦合较大的情况下,电子自旋取向不同的态受到自旋轨道耦合的激发,从而使电子具有跃迁到此激发态的概率,因此电子可能在不同的自旋状态下跃迁.这使得哈密顿量不再具有各向同性.
DM 相互作用的交换参数Dij是矢量,其方向为
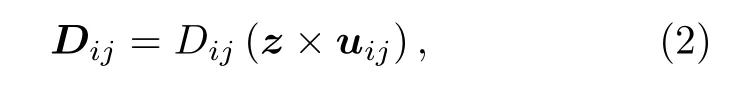
其中uij是原子i指向原子j方向的单位基矢,z是具有自旋轨道耦合原子垂直于原子i与原子j连线方向的基矢.Dij参数具有反对称性质,即Dij=—Dji.因此在对称性较高的晶体中,DM 相互作用会抵消.
DM 相互作用是产生斯格明子的重要机理.斯格明子是一种受拓扑保护的稳定的自旋结构,2009年德国慕尼黑大学首次在实验上观测到斯格明子的存在[13],这吸引了大量学者致力于斯格明子的研究.斯格明子的半径在纳米量级,可以用很小的电流操控,因此可以作为数据存储以及制造自旋电子器件的良好材料.
根据自旋结构的不同,斯格明子主要分为布洛赫型斯格明子和奈尔型斯格明子.奈尔型斯格明子大多出现在铁磁材料和重金属材料形成的多层膜界面上.重金属的原子间有较大的自旋轨道耦合,因此界面上存在较强的DMI,且DMI 矢量的方向垂直于膜界面,从而使得界面上出现稳定的奈尔型斯格明子.奈尔型斯格明子的结构如图1 所示,该结构每个原子的自旋磁矩方向沿着径向缓慢旋转,直到边界上的原子与中心原子的自旋磁矩方向相反.

图1 奈尔型斯格明子的磁矩结构示意图Fig.1.Spin configuration of Néel skyrmions.
通过第一性原理的计算,可以得到二维磁性晶格在不同的磁矩结构下的能量,而不同磁结构下能量与交换参数的关系可以通过磁相互作用的哈密顿量来表示.目前,国内大多数对于磁性材料交换参数的计算[14-16],是采用能量映射的方法.该方法的大致步骤为[17]:1)确定好系统中磁性原子的结构,预设N个磁矩的分布;2)利用VASP 程序计算出这N种磁矩分布的能量,并两两相减获得N -1个相互独立的能量差;3)根据海森伯和DM 模型表达出这N种磁矩分布的能量表达式,并写出第二步的能量差在这两种模型下与交换参数的关系式;4)通过最小二乘法拟合出交换参数的大小.在能量映射方法中,交换参数的大小容易受到磁矩结构的影响,因此,磁矩结构的构建具有十分重要的作用.在计算DM 交换参数的时候,还需要考虑具有非共线的磁矩结构,否则DM 交换参数的计算结果在共线的磁矩结构下可能是0.
在目前使用的能量映射方法的过程中,仍存在着诸多的缺点.首先,对于二维磁性晶格来说,该方法能够构建的磁矩结构不够多,因此容易出现过度拟合的情况,导致拟合的各近邻磁交换参量脱离了实际意义;其次,由于磁结构构建的有限性,各结构之间没有连续的关联性,因此,其中某些结构的能量计算出现错误,或者由于磁矩本身的易变性导致能量发生了无意义的改变,这些都不能从计算值中合理地剔除出去而参与了磁交换参量的拟合,这更加剧了交换参量拟合的失常;第三,这种方法不能考虑太多近邻数,近邻数越多,需要构建的磁构型数(N)就越多,这将大大地增加计算量;最后,对于金属体系,不同自旋结构下同一近邻的相互作用往往并不一样.因此,需要新的计算方法来获取各近邻参量.
2002年,Marsman 和Hafner[18]首次通过广义布洛赫条件计算γ-Fe 自旋螺旋色散关系,从而得到了各近邻参数J.在广义布洛赫条件下[19,20],磁性原子的磁矩是呈周期性变化的.二维磁性体系每个磁性原子的磁矩在同一平面内.以倒格基矢来表示自旋波的波矢k=q1b1+q2b2,原胞基矢来表示每一个格点的位置R=ma1+na2.则两个磁性原子间的磁矩关系为
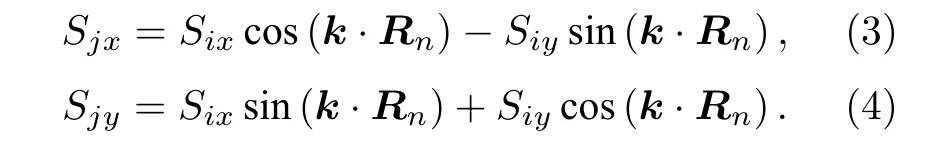
由(3)式和(4)式可知,根据波失的不同,可以构造多个不同的磁矩结构.当k的值在倒格基矢下表示为(0,0,0)时,各个原子的磁矩方向相同,因此代表着铁磁的结构;当k值表示为(0.5,0.5,0),j原子的磁矩与i原子的磁矩不再平行,从而代表了某个特定的反铁磁的结构.同理可得,其他的k值代表着更多的磁性结构.因此通过广义布洛赫的条件可以便捷且有规律地构造大量的磁构型.
本文将利用广义布洛赫条件,推导在海森伯和DM 相互作用模型下二维磁性晶格中磁性原子之间的自旋螺旋色散关系,并通过VASP 程序的计算来拟合交换参数.本文计算了4 种磁性晶格结构不同的材料:六角对称结构VSe2,四方对称结构MnB,六角非对称结构MnSTe,原胞中含有两个原子的六角非对称结构Cr2I3Cl3.
2 六角对称结构VSe2
VSe2材料有高载流子浓度、电导率,以及电荷密度波相变行为,在电容器、自旋电子学、太阳能电池等领域具有广阔的应用前景[21].2018年Bollina等通过分子外延技术首次在实验上生长出了单层的VSe2,其在室温下呈现铁磁性.VSe2独特的光电特性吸引了大量的学者进行研究,但部分性质仍在研究中.目前,如何调控VSe2的性质,使其能够更好地应用在光电器件中仍需要投入更多的研究.
2.1 原子结构
VSe2的原子结构如图2 所示.VSe2属于六角晶系,晶格常数为3.46 Å.磁性原子V 层属于六角结构,V 层的上层和下层都是Se 原子层,Se 原子层也是六角结构.因此VSe2属于六角对称结构,体系中不含有DMI.
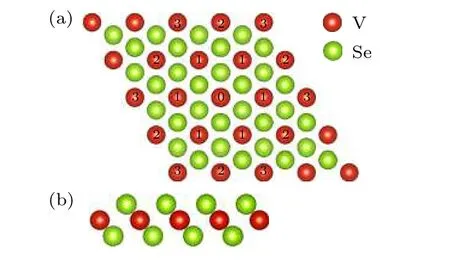
图2 VSe2 原子结构示意图 (a)俯视图;(b)侧视图Fig.2.The view of the lattice structure for VSe2:(a)Top view;(b)side view.
2.2 自旋螺旋色散关系
六角磁性原子结构和各近邻分布的示意图如图3 所示.磁性原子的磁矩变化坐标系在图3(c)中用蓝色的坐标轴表示,而黑色的坐标轴表示二维晶格平面的坐标系.通过设定一个中心原子,计算其他近邻原子与该中心位置的原子之间的相互作用之和,即可确定六角体系中磁矩相互作用情况.
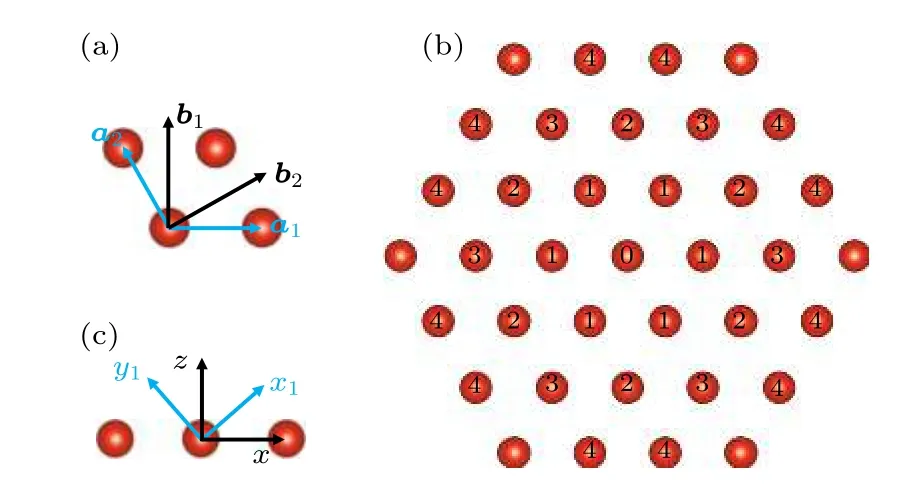
图3 (a)六角结构的原胞基矢和倒格基矢的示意图;(b)六角结构的磁性原子及各近邻原子的分布;(c)磁性原子的磁矩变化坐标系(蓝色坐标轴)以及二维晶格坐标系(黑色坐标轴)示意图Fig.3.(a)The labeled a1 and a2 are basis vectors and b1 and b2 are reciprocal lattice vectors.(b)distribution of neighboring atoms;(c)blue axis and black axis represent the coordinate system of magnetic moment and two dimensional lattice,respectively.
根据(3)式和(4)式,中心原子和其他原子在晶格平面坐标轴3 个方向上的磁矩表达式为

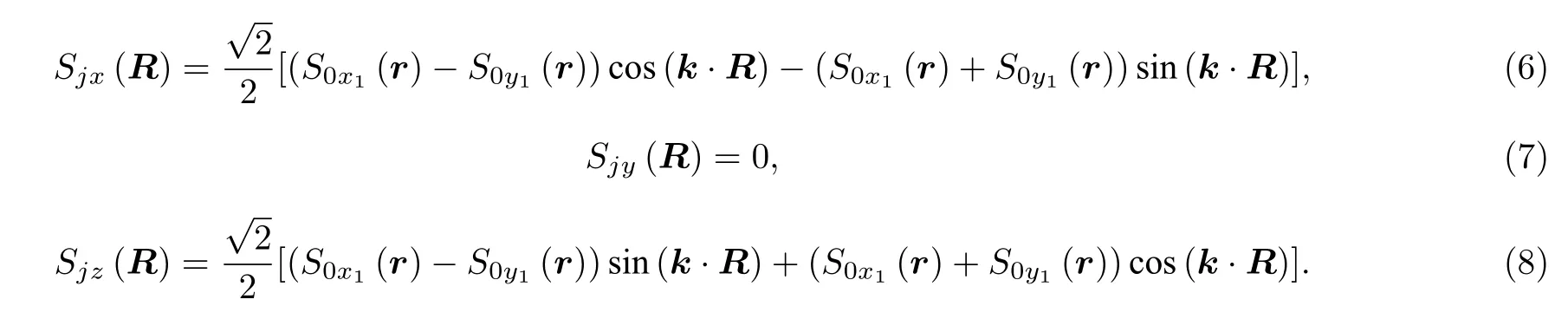
以铁磁态为基态,根据(1)式,六角对称结构的自旋螺旋色散关系为(详细的推导过程见附件1)
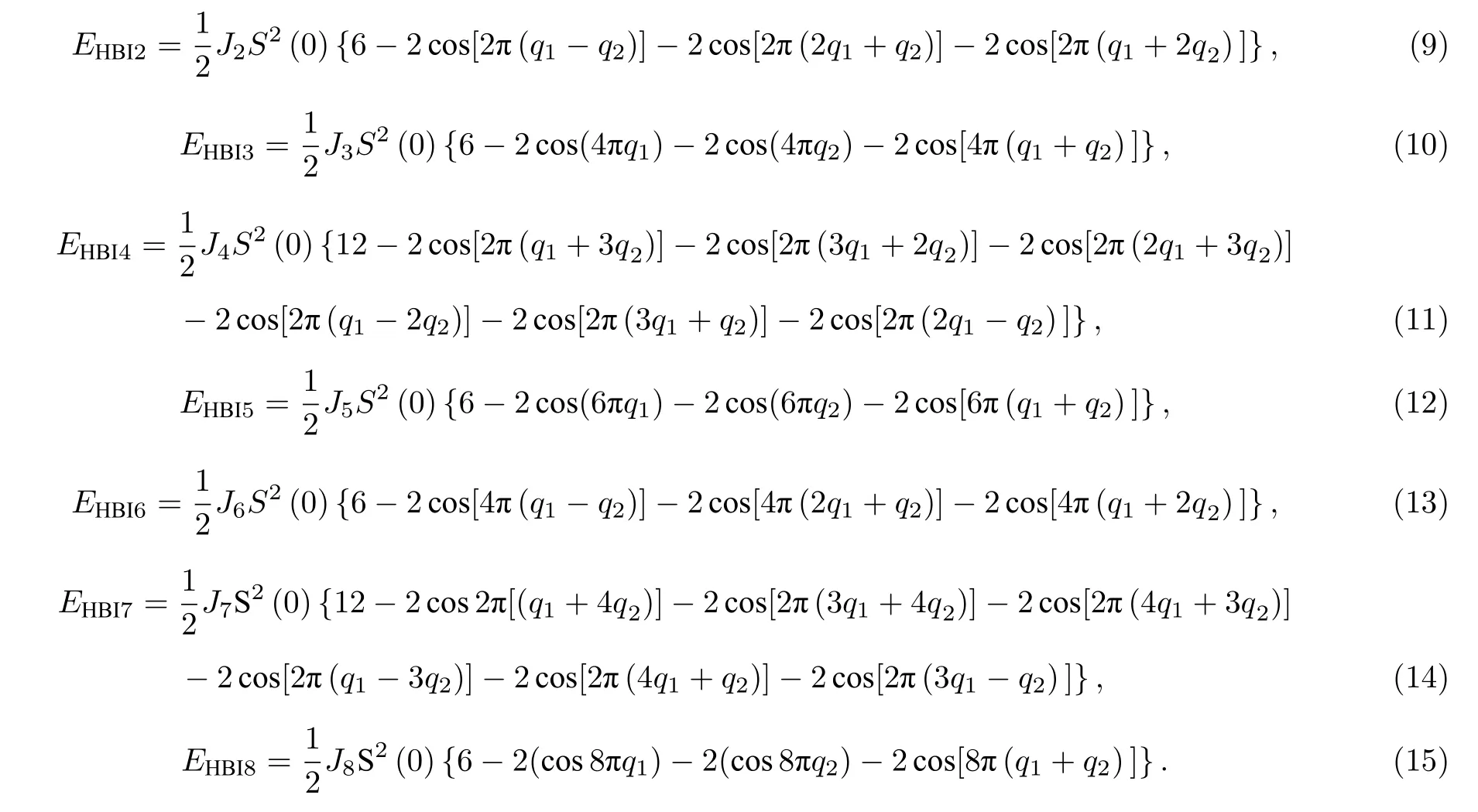
磁性原子的磁交换总能表示为
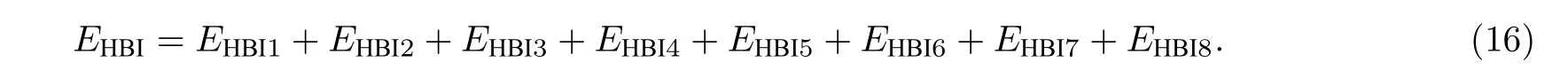
利用程序计算多个不同q参数下磁交换总能EDFT(q),根据(9)式,可以得到不同的波矢q下的海森伯交换作用总能,通过最小二乘法,便可拟合出各近邻的交换参数值.
VSe2材料性质的计算通过VASP 中缀加平面波的方法实现[22,23].平面波的截断能为312 eV,以保证良好的收敛.初始截断能为220 eV,用于延展平面波函数.电子步自洽计算的收敛标准是1×10—6eV.高斯展宽的半径为0.1 eV.对于结构弛豫,所有原子的弛豫收敛标准为10—2eV/Å.二维六角晶格布里渊区的采样点为25×25×1,用HubbardU来描述3 d 电子间的库仑排斥作用,U为3 eV.
计算结果如图4 所示,K,Γ,M为第一布里渊区的高对称点.在磁交换能计算中,选取了K-Γ-M线上以及K-Γ-M-K线所包含区域内的若干个q点,并计算这些q点所代表的磁构型下体系的磁交换总能.图中的离散点为K-Γ-M线上q点的磁交换总能.通过最小二乘法计算得到了交换参数的大小,结果如表1 所列.根据交换参数的值,便可得到不含交换参数的自旋螺旋色散关系EJ(q),图中的实线表示EJ(q)函数在K-Γ-M线上的取值.

表1 VSe2 结构中磁性原子各近邻的海森伯交换参数大小(单位:meV)Table 1.Calculated parameters of Heisenberg exchange J of VSe2,J is considered to the eighth neighbor.(The unit of J is meV).
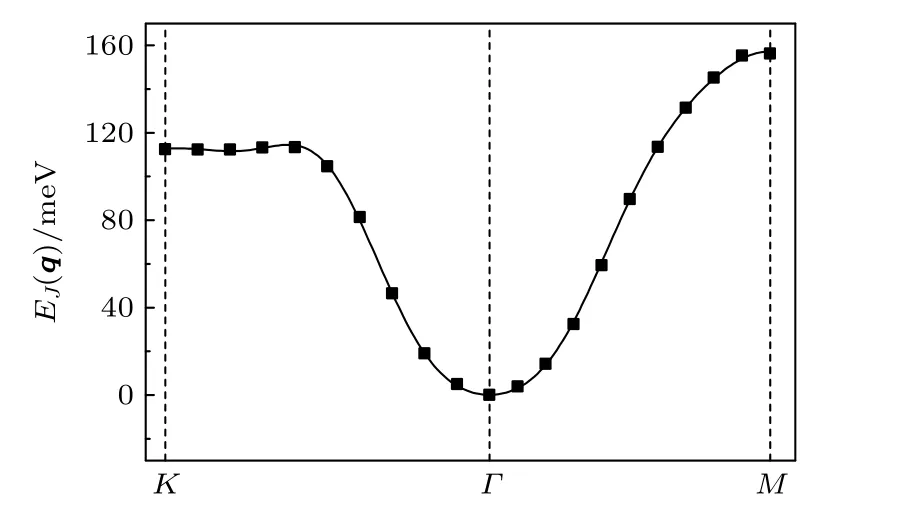
图4 离散点是VSe2 体系通过程序计算得到的自旋螺旋能量色散关系 E(q),其中q 是自旋螺旋的波矢;实线是拟合曲线Fig.4.Scatter symbols are energy dispersion E(q)as a function of the spiral wave vector q calculated by program,lines are fitted ones.
计算结果表明,EJ(q)函数的曲线与程序计算得到的磁交换能(离散点)趋势基本一致,这表明VSe2体系中磁性原子之间的交换作用能基本符合海森伯体系,拟合得到的J值非常合理.在计算的J值中,第一近邻J1和第二近邻J2对体系的磁交换能起到了较大的作用,其值有利于形成铁磁体系;J3有利于形成反铁磁体系,但由于其值太小,没能改变体系的磁基态,因此该体系属于铁磁状态.从图4 也可得知,在Γ点磁交换总能是最低的.而在广义布洛赫条件下,Γ点代表的磁构型是各个原子的磁矩方向保持一致.因此该体系的稳定磁状态为铁磁.
3 四方对称结构MnB
2017年,Farooq 等[24]通过结构预测算法,提出了具有p4mmm对称的MnB 结构,p4mmm对称性使得该结构的物理性质有比较理想的均匀特征.这种MnB 结构中每个原子的内聚能是4.493 eV,与现有的二维(2D)材料的内聚能相当,表明在适当的条件下,可以在实验上制备出单层的MnB 结构.同时,该结构也具有热学稳定性,Farooq 等[24]的分子动力学模拟结果表明,即使在600 K 的高温下,MnB 的结构也没有发生大幅度的变化.在MnB 结构的二维平面内,x和y方向的杨氏模量都是100 N/m,表明了该结构具有力学稳定性.总而言之,p4mmm对称的MnB 结构不仅能够通过适当的实验方法制备出来,还能在室温下稳定存在.
在没有应力的情况下,MnB 的费米能级具有一个狄拉克环,通过增加适当的应力,能够使MnB结构的拓扑性质和磁基态发生相变[25],这一性质使其为数据传输领域提供较大的帮助,对于开发纳米结构的自旋电子设备具有重要的意义.
3.1 原子结构
MnB 结构的俯视图和侧视图如图5 所示,其晶格常数的计算结果为2.85Å.MnB 属于底心立方结构,每一层的原子结构相同,表示MnB 具有空间反演对称,磁性原子Mn 之间不存在DM 交换作用.MnB 二维晶格体系属于对称的四方结构.
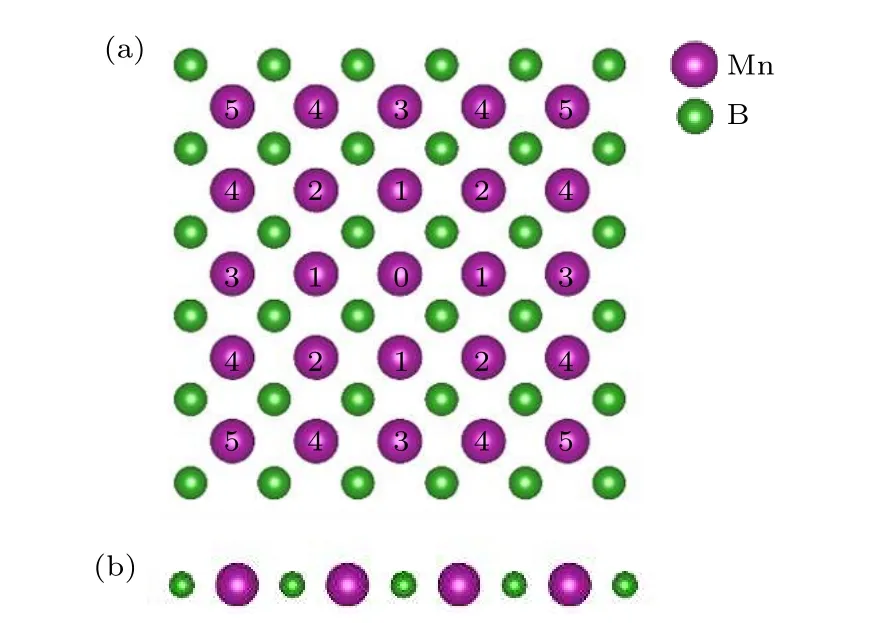
图5 MnB 原子结构示意图 (a)俯视图;(b)侧视图Fig.5.Structure of MnB:(a)Vertical view;(b)side view.
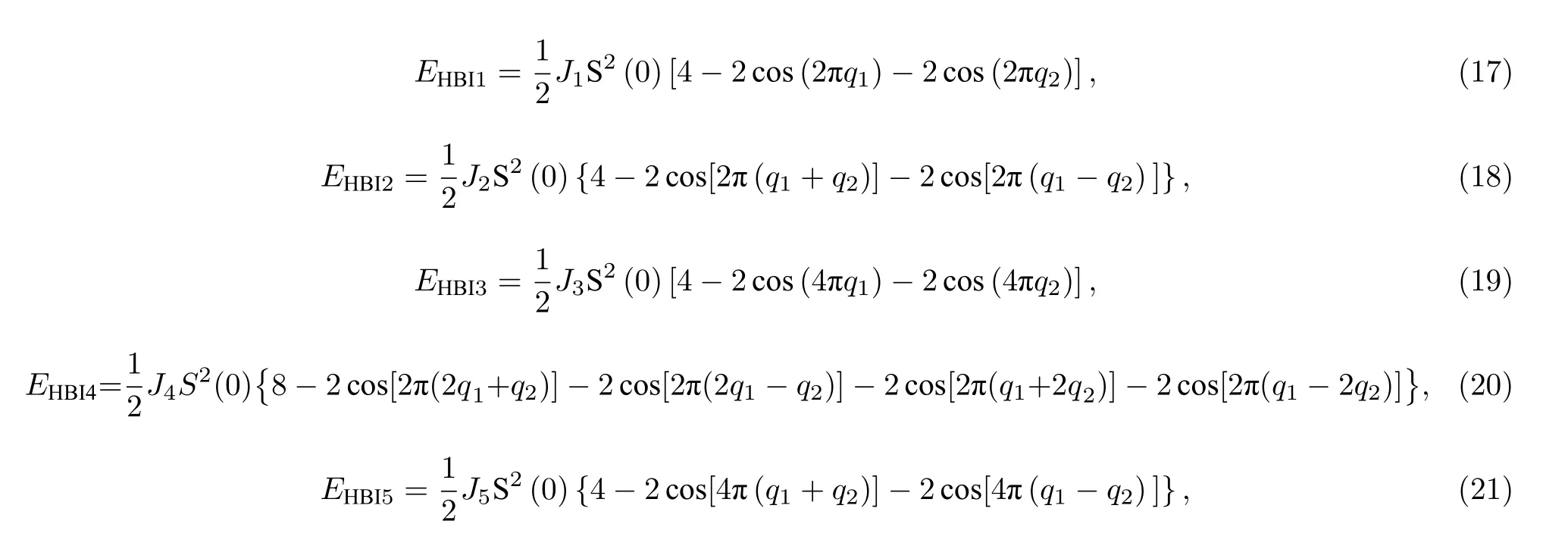
3.2 自旋螺旋色散关系
与六角结构的推导方法类似,得到四角对称结构二维磁性晶格的自旋螺旋色散关系为:四角结构在HBI 下的磁交换作用总能量满足(16)式.
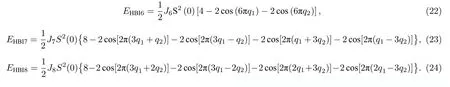
本次计算同样利用VASP 中缀加平面波的方法实现.平面波的截断能为419 eV,初始截断能为319 eV.电子步自洽计算的收敛标准是1×10—6eV.高斯展宽的半径为0.02 eV.对于结构弛豫,所有原子的弛豫收敛条件为10—2eV/Å.二维四方晶格布里渊区的采样点为25×25×1.用HubbardU来描述3 d 电子间的库仑排斥作用,U选为3 和5 eV.
计算结果如图6 所示,离散的点表示程序计算的磁交换总能在不同波矢下的值,通过在HBI 下四方结构的自旋螺旋色散关系和最小二乘法拟合,得到了各近邻交换参数的大小(表2),从结果可知,对系统的磁交换能有较大影响的近邻参数为J1,J2,J3,J4.第一和第二近邻对磁性交换作用能的贡献占主要部分,其中第二近邻的贡献最大;第三近邻和第四近邻虽是负的,但由于其值比第一和第二近邻小了一个数量级,不足以对磁矩方向的分布有大的影响.从图6 可知,在q为0 的地方,即Γ点,MnB 结构的磁性交换作用能最低,因此所有原子的磁矩方向一致,MnB 结构呈现铁磁性.

表2 MnB 结构(U =3 eV)中磁性原子各近邻的海森伯交换参数大小(单位:meV)Table 2.Calculated parameters of Heisenberg exchange J of MnB,J is considered to the eighth neighbor.(The unit of J is meV).
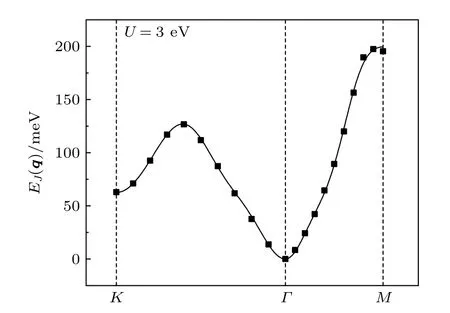
图6 离散点是MnB 体系通过程序计算得到的自旋螺旋能量色散关系 E(q),其中q 是自旋螺旋的波矢;实线是拟合曲线Fig.6.Scatter symbols are energy dispersion E(q)of MnB as a function of the spiral wave vector q calculated by program,lines are fitted ones.
当U增加到5 eV 时,MnB 的磁交换作用能变小,而磁性基态也从铁磁状态变为反铁磁状态,这主要是由第一近邻的值变为负数(—10.01 meV)引起的.如图7 所示,MnB 的磁交换总能在K点的时候最低,K点的q值为(0.5,0.5,0).根据广义布洛赫条件,每个磁性原子的磁矩与其最近邻的磁矩相反.因此MnB 体系呈现反铁磁的特性.
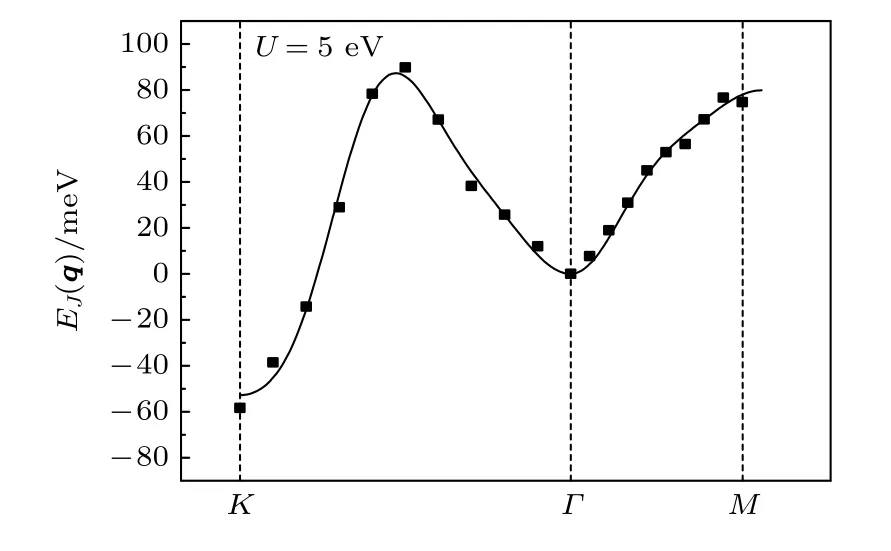
图7 离散点是MnB 体系通过程序计算得到的自旋螺旋能量色散关系 E(q),其中q 是自旋螺旋的波矢;实线是拟合曲线Fig.7.Scatter symbols are energy dispersion E(q)of MnB as a function of the spiral wave vector q calculated by program,lines are fitted ones.
4 六角非对称结构MnSTe
在二维磁性材料中,存在DM 相互作用的关键要素是材料具有空间反演对称的结构.2017年,Zhang 等[26]通过控制MoS2材料的生长,利用Se原子取代顶层的S 原子,成功制备了具有空间反演对称破缺的二维Janus 单层材料MoSSe.受此启发,相同结构但含有磁性原子的Janus 材料MnSTe也得到了理论的研究[27],结果表明,MnSTe 能够稳定存在,且具有高度自旋极化和较高的居里温度,这表明二维磁性材料MnSTe 具有潜在的应用价值.
4.1 原子结构
MnSTe 的原子结构如图8 所示,其晶格常数的计算值为3.56 Å.由图8 可知,MnSTe 属于六角晶系,原胞内有3 层原子,中间层是磁性原子Mn,两边分别为S 原子和Te 原子,3 层原子都属于六角结构,因此MnSTe 结构具有空间反演对称破缺,属于六角非对称结构.
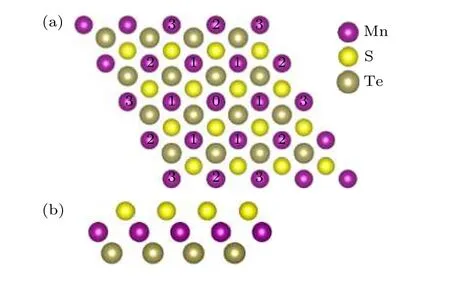
图8 MnSTe 原子结构示意图 (a)俯视图;(b)侧视图Fig.8.Atomic structure of MnSTe:(a)Vertical view;(b)side view.
4.2 自旋螺旋色散关系
相比于六角对称结构,六角非对称结构需要再增加一项DM 相互作用.与六角对称结构的推导方法类似,六角结构在DM 相互作用下磁交换作 用能的自旋螺旋色散关系(考虑到第四近邻)为
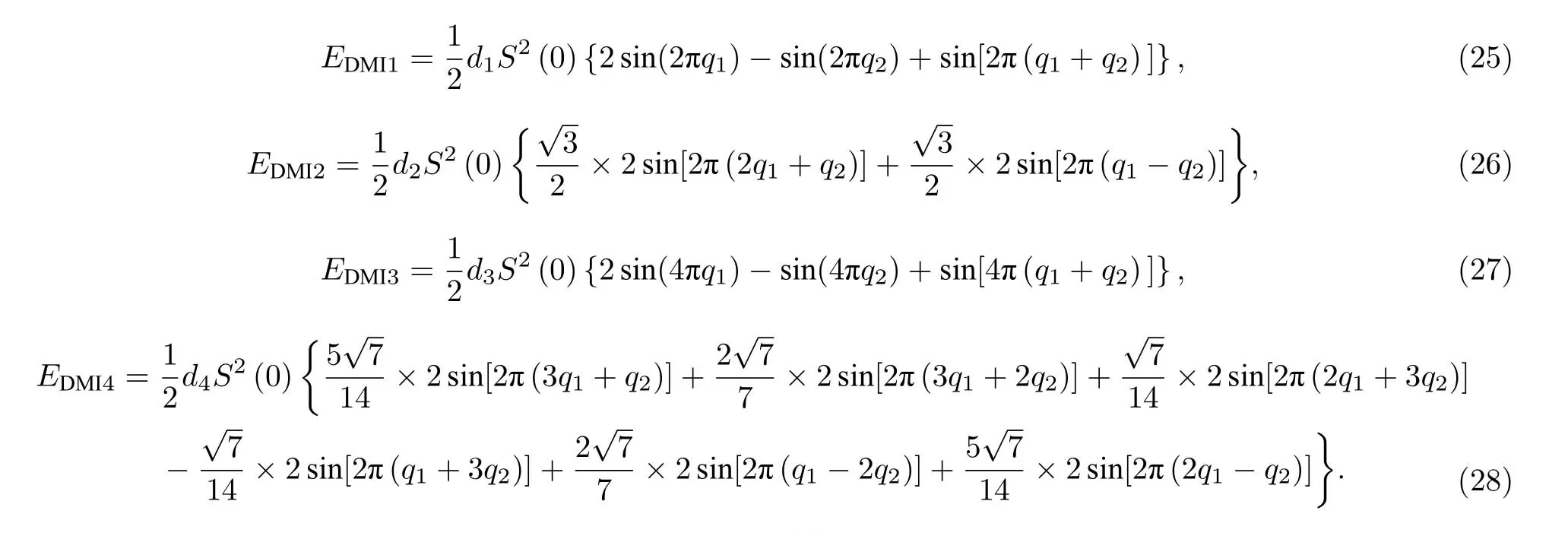
DM 相互作用下的磁交换总能为
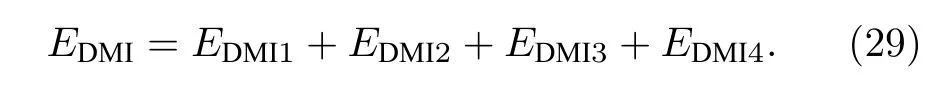
对于含有DM 交换作用的系统,程序计算的磁交换作用总能主要来自于海森伯相互作用和DM相互作用的贡献,即
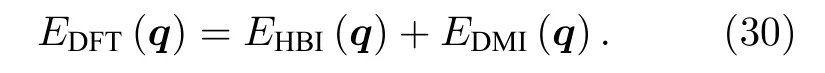
由于DM 相互作用下的磁总能是关于q的奇函数,而海森伯作用下磁总能是关于q的偶函数.即
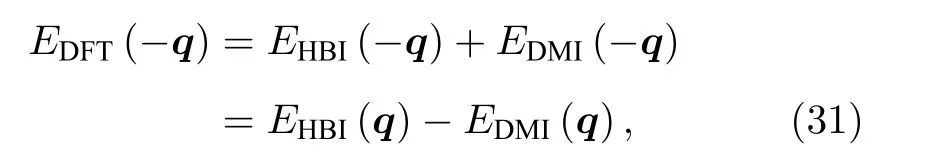
因此DM 作用的能量可以表示为
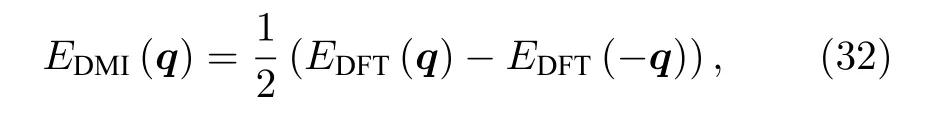
海森堡相互作用的能量可以表示为
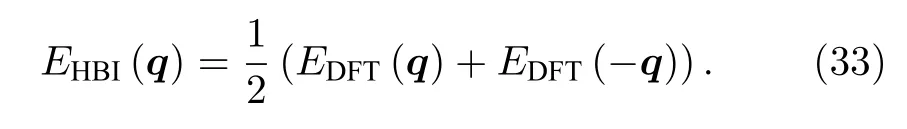
通过最小二乘法,即可通过VASP 程序拟合得到海森伯和DM 交换参数的大小.
本次计算的平面波的截断能为380 eV,初始截断能为280 eV.电子步自洽计算的收敛标准是1×10—6eV.高斯展宽的半径为0.02 eV.对于结构弛豫,所有原子的弛豫收敛标准为10—2eV/Å.二维六角晶格布里渊区的采样点为25×25×1.
MnSTe 的计算结果表明(图9),Γ点的能量最低,因此体系呈现铁磁状态.在各海森伯交换参数中(表3),对磁交换作用能影响较大的参数是J1,J2,J3,J4,J5,其中第三近邻的值为7.29 meV,是所有交换参数中的最大值.J6—J8的参数值很小,因此其对海森伯能量的贡献可以忽略.J4和J5的值虽然是负值,但是数值远小于J1—J3,因此引起的体系磁矩方向的改变量很小,最终导致体系属于铁磁状态.

表3 MnSTe 结构中磁性原子各近邻的海森伯交换参数大小(单位:meV)Table 3.Calculated parameters of Heisenberg exchange J of MnSTe,J is considered to the eighth neighbor.(The unit of J is meV).
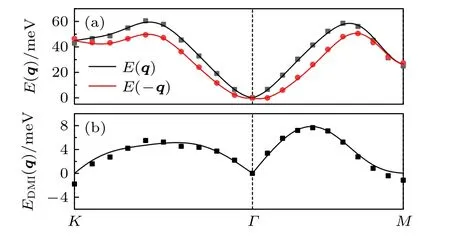
图9 (a)离散点是MnSTe 体系通过程序计算得到的自旋螺旋能量色散关系 E(q)和E(—q),其中q 是自旋螺旋的波矢,实线是拟合曲线;(b)离散点是MnSTe 体系通过程序计算的E(q)与E(—q)之间的能量差EDMI(q),实线是拟合曲线Fig.9.(a)Scatter symbols are energy dispersion E(q)and E(—q)of MnSTe as a function of the spiral wave vector q calculated by program,lines are fitted ones;(b)scatter symbols are EDMI(q)which means the difference between E(q)and E(—q),lines are fitted ones.
在各DM 交换作用参数中(表4),与海森伯交换参数相比,DM 参数不可忽略.d1的值(5.64 meV)对海森伯能量的贡献为最大,且其值与J1相当,因此MnSTe 中DM 相互作用具有相对重要的角色.

表4 MnSTe 结构中磁性原子各近邻的DM 交换参数大小(单位:meV)Table 4.Calculated parameters of DM exchange d of MnSTe,d is considered to the forth neighbor.(The unit of d is meV).
5 原胞中含有两个磁性原子的非对称六角结构Cr2I3Cl3
如前文所述,目前成功制造Janus 单层MoSSe的方法是控制MoS2的生长,用Se 原子替代顶层的S 原子.这一方法也预示了实验上生长Cr2I3Cl3的可能性.Cr2I3Cl3不仅有较强的DMI,其磁各向异性也较小,这些性质有助于体系形成稳定的螺旋磁体或斯格明子[28],因此Cr2I3Cl3材料也具备一定的应用价值.
5.1 原子结构
Cr2I3Cl3的原子结构示意图如图10 所示,属于六角晶系,晶格常数为6.66Å.原胞内Cr2I3Cl3原胞内共有3 层原子,中间层是具有磁性的Cr 原子,Cl 原子层和I 原子层属于原胞含有3 个原子的六角结构.在一个晶体原胞中,以原胞基矢为单位,两个磁性原子Cr 的相对坐标分别为(0,0),(1/3,2/3).Cr2I3Cl3同样存在DM 相互作用,因此其磁性原子的磁交换总能需要考虑HBI 和DMI的共同作用.
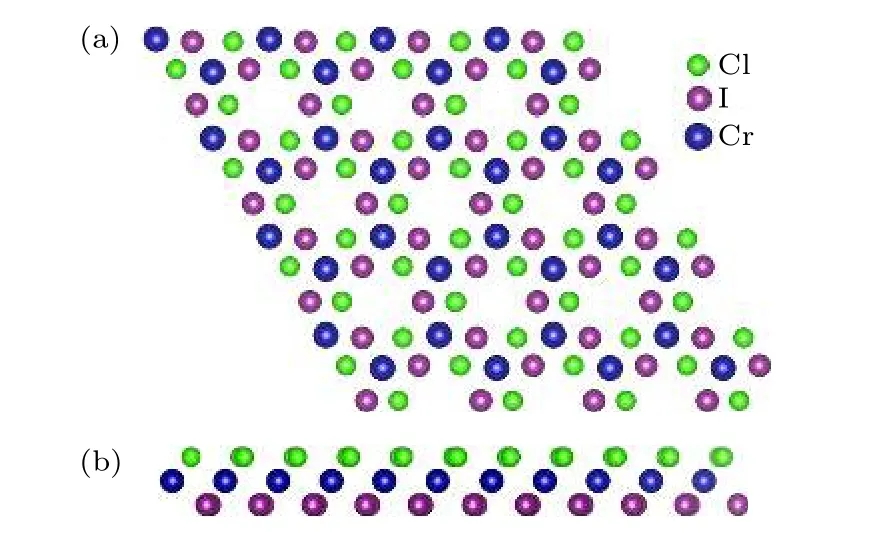
图10 Cr2I3Cl3 原子结构示意图 (a)俯视图;(b)侧视图Fig.10.Atomic structure of Cr2I3Cl3:(a)Vertical view;(b)side view.
5.2 自旋螺旋色散关系
Cr2I3Cl3原子结构在HBI 下各近邻的自旋螺旋色散关系(考虑到第六近邻)为
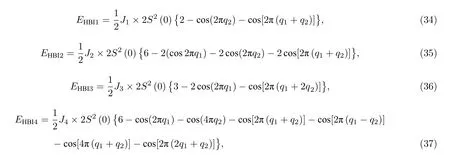
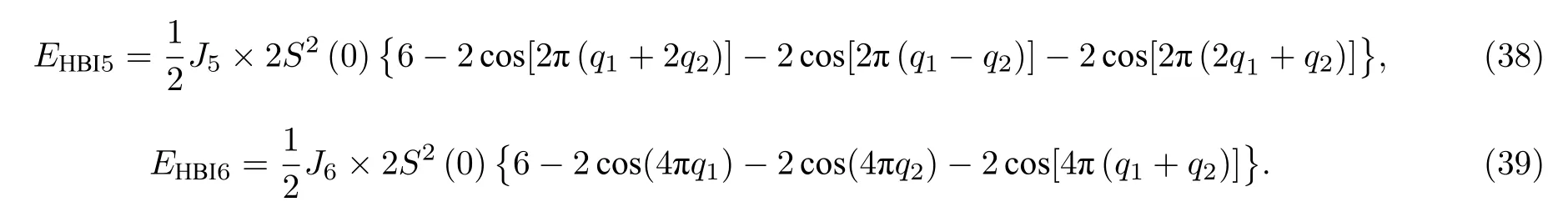
该结构在HBI 下磁交换总能满足(19)式.
在DMI 下各近邻的自旋螺旋色散关系(考虑到第四近邻)为
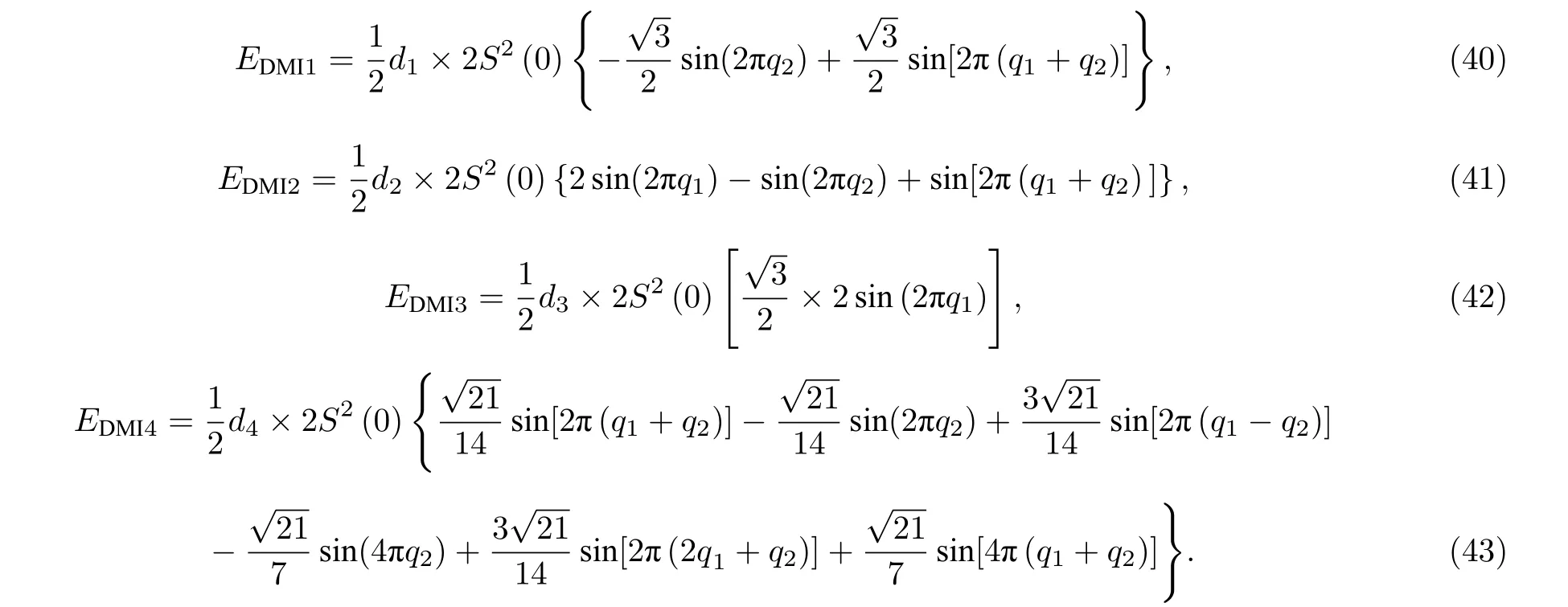
在DMI 下磁交换总能满足(29)式.
本次计算的平面波的截断能为380 eV,初始截断能为280 eV.电子步自洽计算的收敛标准是1×10—6eV.高斯展宽的半径为0.02 eV.对于结构弛豫,所有原子的弛豫收敛标准为10—2/Å.二维六角晶格布里渊区的采样点为9×9×1.
Cr2I3Cl3的计算结果如图11 所示,在Γ点体系的磁交换总能量最低,因此Cr2I3Cl3稳定的磁分布为铁磁状态.体系磁交换总能中海森伯相互作用占据了重要的部分,而DM 相互作用比较微弱.DM 相互作用在K点的能量最低,因此DMI 更容易形成反铁磁的状态,但相对于海森伯交换参数来说,DM 交换参数过低,没能改变体系的总磁矩分布.
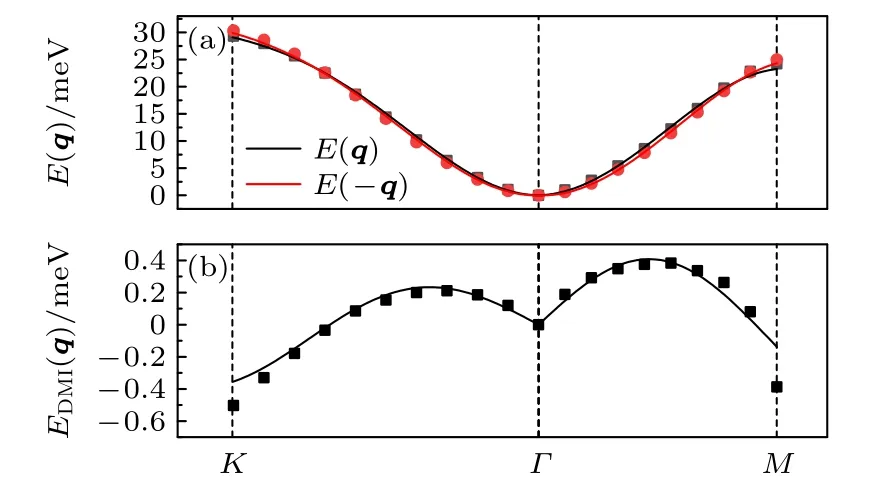
图11 (a)离散点是Cr2I3Cl3 体系通过程序计算得到的自旋螺旋能量色散关系 E(q)和E(—q),其中q 是自旋螺旋的波矢,实线是拟合曲线;(b)离散点是Cr2I3Cl3 体系通过程序计算的E(q)与E(—q)之间的能量差EDMI(q),实线是拟合曲线Fig.11.(a)Scatter symbols are energy dispersion E(q)and E(—q)of Cr2I3Cl3 as a function of the spiral wave vector q calculated by program,lines are fitted ones;(b)scatter symbols are EDMI(q)which means the difference between E(q)and E(—q),lines are fitted ones.
在Cr2I3Cl3的各近邻参数(表5 和表6)中,第一近邻的海森伯交换作用最为重要,相对于海森伯的交换参数,DMI 参数非常小,其中第三近邻的交换参数最大.|d1/J1|的值为0.068,而形成斯格明子需要的范围在0.1—0.2 之间[29].因此 C r2I3Cl3并不具备形成斯格明子的条件.

表5 Cr2I3Cl3 结构中磁性原子各近邻的海森伯交换参数大小(单位:meV)Table 5.Calculated parameters of Heisenberg exchange J of Cr2I3Cl3,J is considered to the eighth neighbor.(The unit of J is meV).

表6 Cr2I3Cl3 结构中磁性原子各近邻的DM 交换参数大小(单位:meV)Table 6.Calculated parameters of DM exchange d of Cr2I3Cl3,d is considered to the forth neighbor.(The unit of d is meV).
6 结论
根据广义布洛赫条件,利用第一性原理软件VASP计算了磁相互作用.根据四角结构、六角结构以及原胞中含有两个原子的六角结构的自旋螺旋色散关系式,对 V Se2,MnB,MnSTe 以及 Cr2I3Cl3四种结构的磁相互作用参数进行了计算和讨论.
V Se2和MnB 的磁交换作用能都不包含DMI.因此海森伯相互作用在磁交换作用能中占据了重要的作用.其中 V Se2属于六角结构,MnB 属于四角结构.计算结果表明,在U=3 eV 的情况下,VSe2和MnB 属于铁磁体.但当U升高至5 eV 时,MnB的磁交换作用能下降,并且呈现反铁磁的状态.
MnSTe 和Cr2I3Cl3都属于Janus 单层,他们的结构相似,都是磁性原子层处于不同的非磁性原子层中间.因此MnSTe 和Cr2I3Cl3都具备空间反演对称条件,体系中含有DM 相互作用.其中MnSTe是六角结构,Cr2I3Cl3的原胞基矢也呈现六角结构,但其原胞中含有两个磁性原子,相对坐标分别为(0,0)和(1/3,2/3).计算结果表明,MnSTe 和Cr2I3Cl3都属于铁磁态.MnSTe 的DM 作用参数较大,与海森伯交换作用参数相当.而Cr2I3Cl3的DM 交换作用参数较小,不足以形成斯格明子相.
