一例含阴离子水簇的钴配合物的合成、结构及理论研究
2022-01-13李慧玲李荣春魏荣敏
李慧玲,杨 敏,孙 金,李荣春,魏荣敏
(德州学院化学化工学院,山东省功能材料与配位化学高校重点实验室,德州 253023)
0 引 言
水簇是指三个及以上水分子通过氢键作用相互连接而形成的水的聚合体。小型水簇因其特殊的不规则性质及其在无机材料、超分子体系、生物分子及化学工程上的重要作用,引起了科学家的极大兴趣,人们对其进行了广泛的研究[1-4]。这不仅为人们研究宏观意义上的水提供帮助,而且有助于人们了解与蛋白质分子有关的水分子的结构。
金属配合物由于其在光学、磁学和电学等方面具有潜在的应用价值,受到了广泛研究[5-7]。研究发现,配合物晶体中位于主体框架结构间的水簇是表征从孤立水分子到大量水的结构变化及键合机理的有效方式,晶体中配合物的主体框架结构可以为水簇提供有效的存在环境。因此,越来越多的包含三聚体[8]、四聚体[9-10]、五聚体[11]、六聚体[12]及八聚体[13-14]等水簇的配合物晶体被广泛研究。其中,环状水四聚体对于理解液态水和冰的双结构模型起着至关重要的作用[1],根据该模型,立方的水八聚体解离成环状水四聚体似乎是在液态水中发生的关键过程。基于此,本文设计合成了阴离子水簇赖以存在的超分子配合物[Co(2,2-bipy)2(N3)2](N3)0.5Cl0.5·2H2O,并培养了其单晶体,采用X射线单晶衍射、红外光谱等对其结构进行了一系列表征,得到了一个包含环状水四聚体的阴离子水簇[(H2O)4(N3)Cl]2-。基于密度泛函理论对配合物的结构、单点能、原子偶极矩校正的Hirshfeld布居(ADCH)原子电荷等进行了理论研究。
1 实 验
1.1 主要仪器与试剂
仪器:单晶X射线衍射使用Bruker Smart CCD 单晶衍射仪测定;红外光谱使用Bruker Vector 22 型红外光谱仪测定;元素分析使用Perkin Elmer 240C型元素分析仪测定。
试剂:六水合氯化钴、叠氮化钠、2,2-联吡啶、乙醇。实验过程中所使用的药品和试剂没有经过进一步的纯化。
1.2 配合物的合成
将CoCl2·6H2O(1.5 mmol,0.36 g)溶于40 mL蒸馏水中,边搅拌边逐滴加入NaN3(3 mmol,0.20 g)水溶液10 mL,溶液搅拌反应1 h后,逐滴加入2,2-联吡啶(2 mmol,0.31 g)的乙醇溶液10 mL,继续搅拌反应2 h后,于暗处静置缓慢挥发,约一周后得到暗红色块状晶体1(0.52 g,产率为65%,以金属Co为基准)。
配合物分子式为:C20H20Cl0.5N11.5O2Co(Mr=530.13),理论计算得出的C、H、N含量分别为:51.28%,5.19%,29.90%;元素分析仪实测C、H、N含量分别为:51.29%,5.17%,29.88%。红外光谱(KBr,ν/cm-1)呈现的主要特征峰为:ν(O—H)3 396.04s;ν(C—H)3 026.33m;ν(N=N)2 031.26vs,2 018.9vs;ν(C=C,C=N)1 650.7w,1 604.96m,1 560.68w,1 497.92w,1 469.63m;ν(C-H)772.17s,727.54m。
1.3 晶体结构解析
选取大小合适的无裂纹、无杂质的高质量晶体,使用Bruker公司的SMART APEX CCD 单晶衍射仪在室温下进行数据收集和还原,使用MoKα(λ=0.071 073 nm)射线,以ω-2θ方式在2.50°~27.86°范围内扫描。衍射数据通过SMART程序收集,并获取相应的晶胞参数,运用SAINT[15]程序对所有衍射点进行精修和数据还原,并使用SADABS[16]程序进行经验吸收校正,然后使用SHELXL-14[17-18]软件包进行晶体结构解析。金属原子坐标由直接法确定,全部非氢原子坐标由Fourier合成及差值电子密度函数修正,氢原子坐标从差值电子密度函数并结合几何分析获得,全部非氢原子坐标、各向异性温度因子经最小二乘法精修至收敛。
1.4 理论计算方法
本文所有计算均基于密度泛函理论(DFT),使用Gaussian 09软件包[19]进行。以X射线单晶衍射法解析得到的晶体结构为基础,选用配合物[Co(2,2-bipy)2(N3)2]+阳离子为计算模型,采用BP86理论方法,对化合物1的低自旋态几何构型(S=0)进行了全优化,并且对优化构型进行了频率分析。其中,C、H、N原子采用6-31g*基组,Co原子采用SDD赝势基组。此外,考虑到量子化学计算模型是分子在气相时孤立状态下的结构,而晶体状态下的分子间存在相互作用,X射线衍射可以准确测出重原子位置,而氢原子依靠的是理论加氢,因此,作为比较,在相同计算水平下,本文对配合物阳离子模型中的氢原子进行了优化。并在此基础上,分别采用BP86、PBE、TPSSH、B3LYP、B3LYP*、M06泛函计算了配合物1高低自旋态的单点能,选用基组为def2-TZVP。
2 结果与讨论
2.1 配合物的单晶结构
选取大小为0.20 mm×0.14 mm×0.20 mm的晶体样品进行单晶X射线衍射分析。部分结晶学数据和结构参数列于表1中,表2为其主要的键长和键角数据。
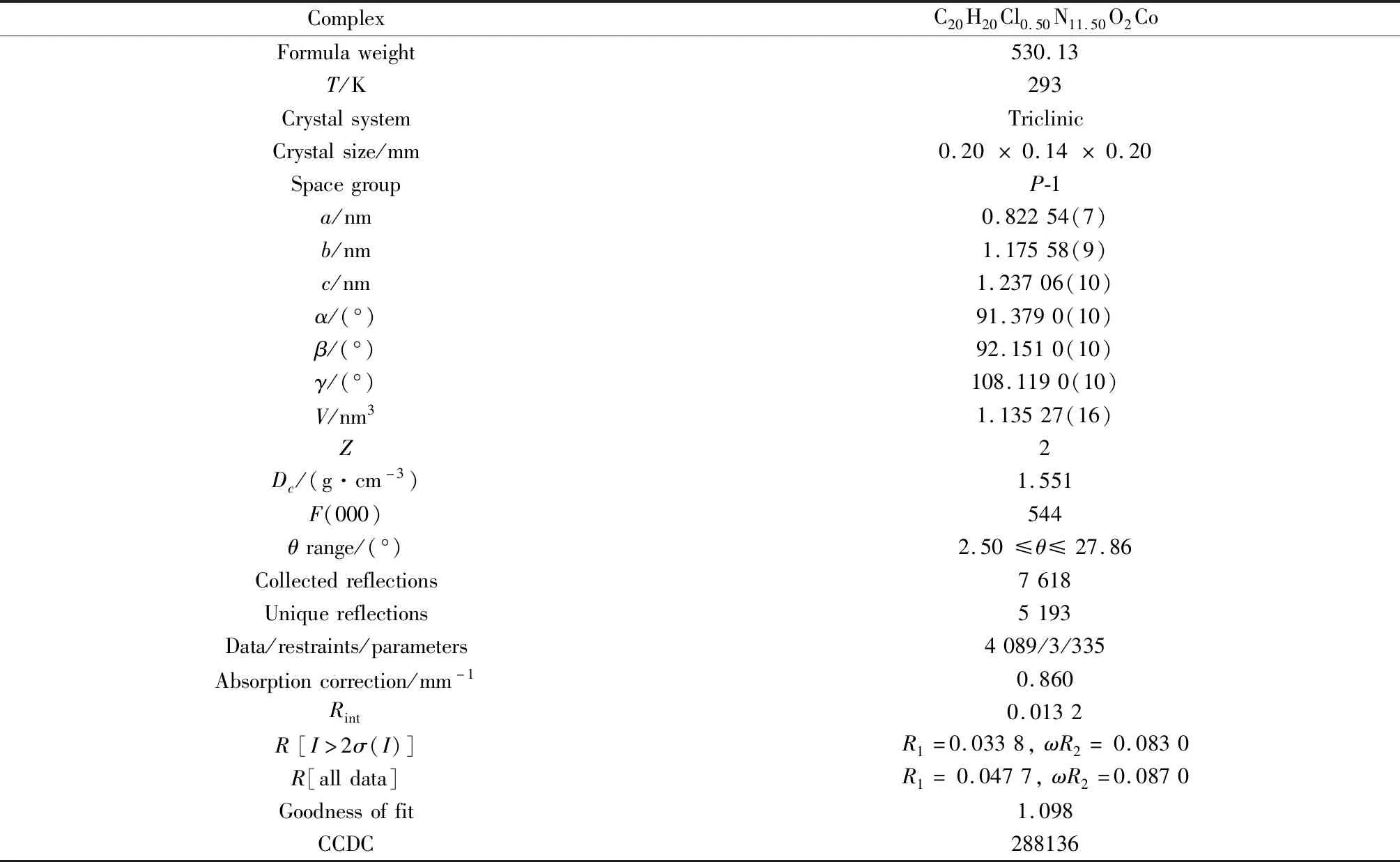
表1 配合物1的部分结晶学数据Table 1 Crystal data and structure refinement for complex 1
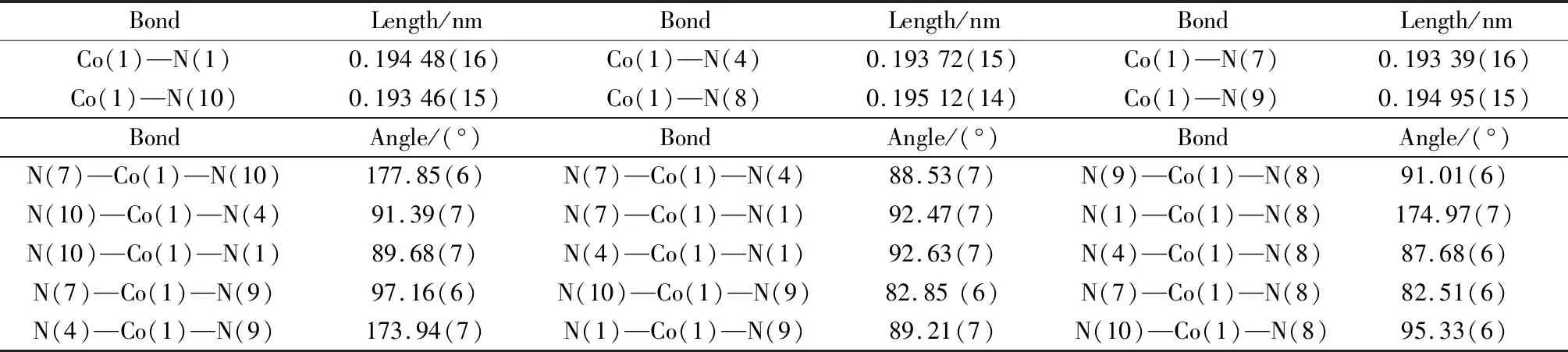
表2 配合物1主要的键长(nm)与键角(°)Table 2 Selected bond lengths (nm)and bond angles (°)for complex 1
单晶结构解析表明,配合物1属于三斜晶系,P-1空间群,其室温下分子结构图如图1(a)所示。从图中可以看出,该配合物每个最小不对称单元包含一个金属钴(Ⅲ)配合物阳离子[Co(2,2-bipy)2(N3)2]+,一个游离的无序Cl-和N3-阴离子,其位置占有率均为50%,此外还包括两个游离的水分子。配合物1中,CoIII离子位于CoN6八面体配位环境中,轴向位置被两个分别来自2,2-联吡啶的N原子(N7,N10)占据,两个叠氮氮原子(N1,N4)与来自2,2-联吡啶配体的另外两个N原子(N8,N9)处于八面体构型的赤道平面位置,轴向Co—N键长稍短于赤道键长,从而形成了压缩的变形八面体配位构型,相应的键长键角数据如表2所示。每个单核配合物阳离子[Co(2,2-bipy)2(N3)2]+通过分子间C—H…π和π…π相互作用,彼此相互连接形成了带状一维超分子结构,如图2所示。
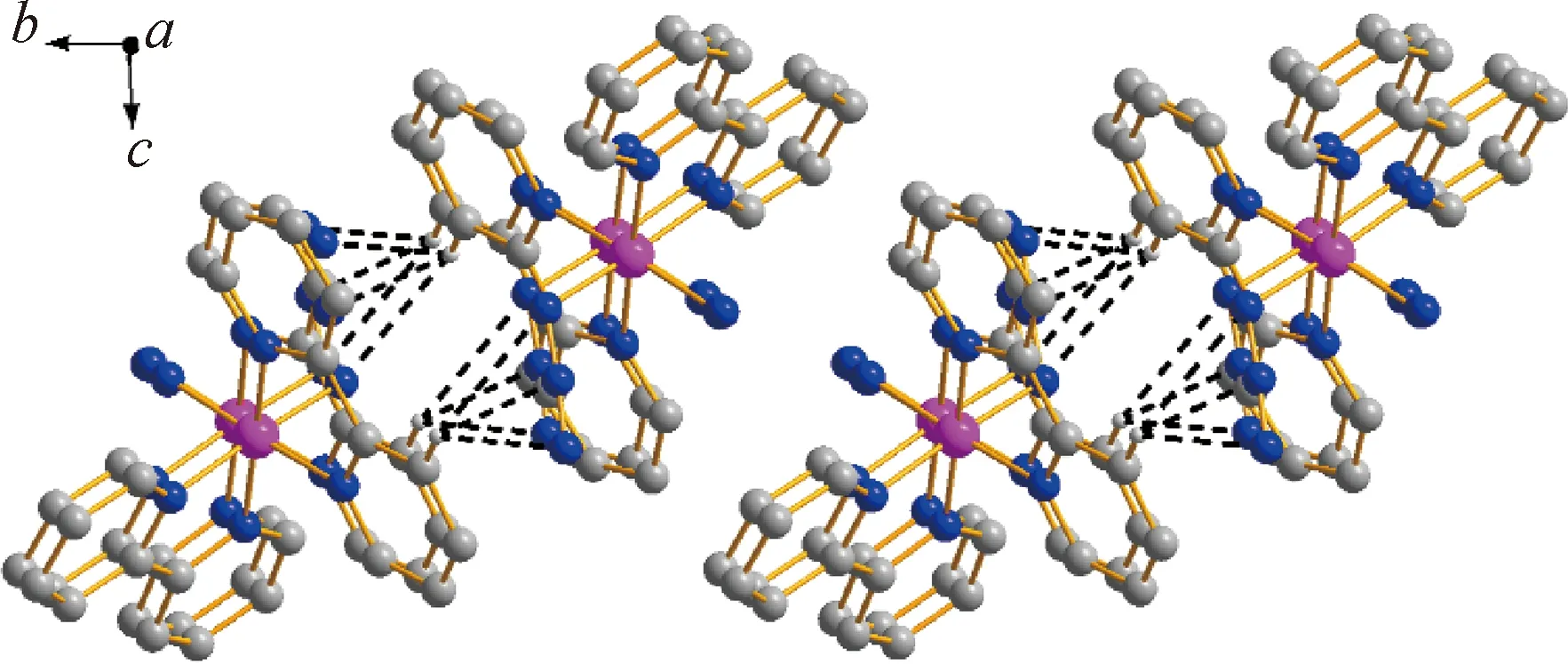
图2 配合物阳离子[Co(2,2-bipy)2(N3)2]+带状超分子结构图(与弱作用无关的氢原子和阴离子水簇均已略去)Fig.2 Ribbon supramolecular structure of [Co(2,2-bipy)2(N3)2]+.(Anionic water clusters and hydrogen atoms without weak interactions have been omitted for clarity)
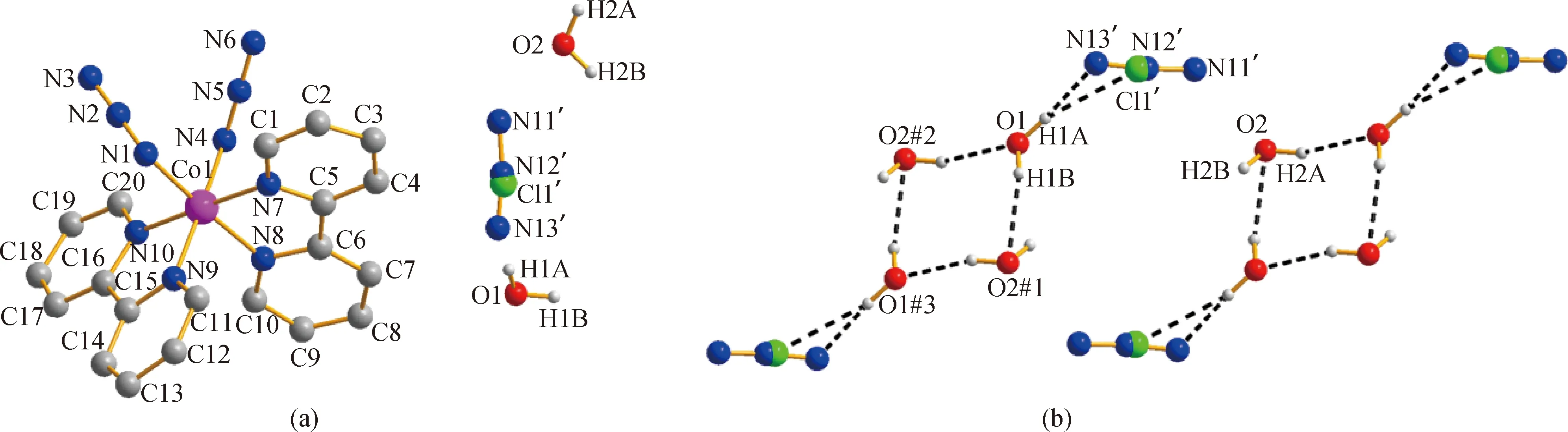
图1 (a)配合物1的分子结构图(2,2-联吡啶的H 原子已略去);(b)阴离子水簇[(H2O)4(N3)Cl]2-结构图(N3-和Cl-离子位置无序)Fig.1 (a)Molecular structure of complex 1 (Hydrogen atoms of 2,2-bipy are omitted for clarity);(b)molecular structure of anion-water cluster and Cl- ions are disordered)

表3 配合物1水簇中氢键的键长(nm)与键角(°)表Table 3 Bond lengths (nm)and angles (°)of hydrogen-bondings for anion water cluster of complex 1
2.2 配合物的理论计算分析
2.2.1 理论模型结构
由X射线单晶衍射得到的晶体结构键长数据可以初步判断,配合物中心金属CoIII离子为低自旋态。因此,本文对配合物1的阳离子[Co(2,2-bipy)2(N3)2]+进行了全构型优化(2S+1=1)。图3为优化得到的配合物的平衡几何构型,频率计算结果显示,简谐振动频率都是正值,说明优化得到的构型是稳定存在的,为能量极小值。表4给出了优化所得平衡几何构型的部分键长和键角值。从图3和表4中可以看出,优化得到的Co—N键长值及相应键角与实验值基本吻合,其间的微小差异主要是因为量子化学计算模型是孤立状态下的气态结构,而实验值是晶体结构,存在分子间作用力和晶格能,因此,在误差范围内,可以认为采用密度泛函理论的BP86/6-31g*+SDD 方法能够得到配合物的准确分子结构。由此可见,当实验难以得到晶体数据时,采用量子化学计算的方法也是一种不错的选择。
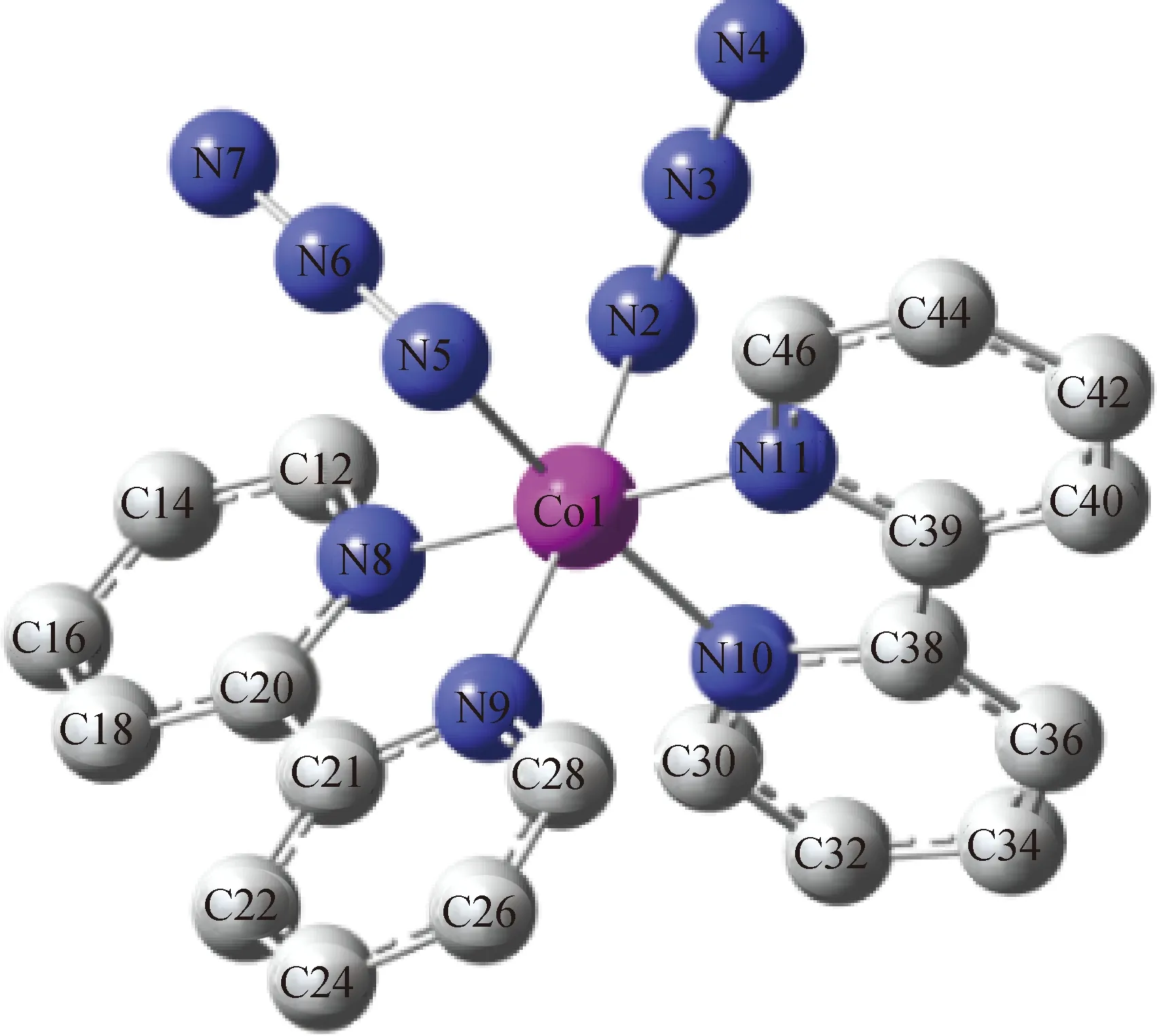
图3 优化得到的[Co(2,2-bipy)2(N3)2]+配合物离子几何构型图(H原子已略去)Fig.3 Optimized [Co(2,2-bipy)2(N3)2]+ ion geometric configuration diagram (H atoms are not shown)
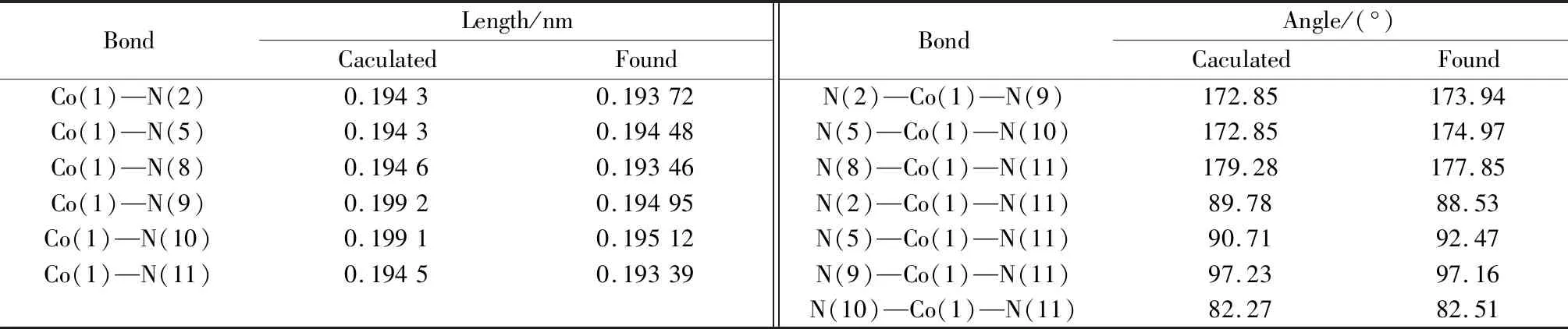
表4 [Co(2,2-bipy)2(N3)2]+配合物离子的全构型优化和实验部分键长、键角Table 4 Band lengths and angles of full geometry optimized and crystal analyzed geometries for complex[Co(2,2-bipy)2(N3)2]+ ion
2.2.2 单点能分析
在对H原子进行限制性优化所得结构的基础上,本文选取不同的泛函,在基组def2-TZVP下,计算了配合物阳离子[Co(2,2-bipy)2(N3)2]+在不同自旋态时的总能量,结果如表5所示。从表中数据可以看出,对于不同泛函,低自旋态单点能均低于高自旋态,由此可以推断出配合物1中CoⅢ为低自旋态,自旋量子数S=0,这与晶体结构所得的键长数据也是吻合的。
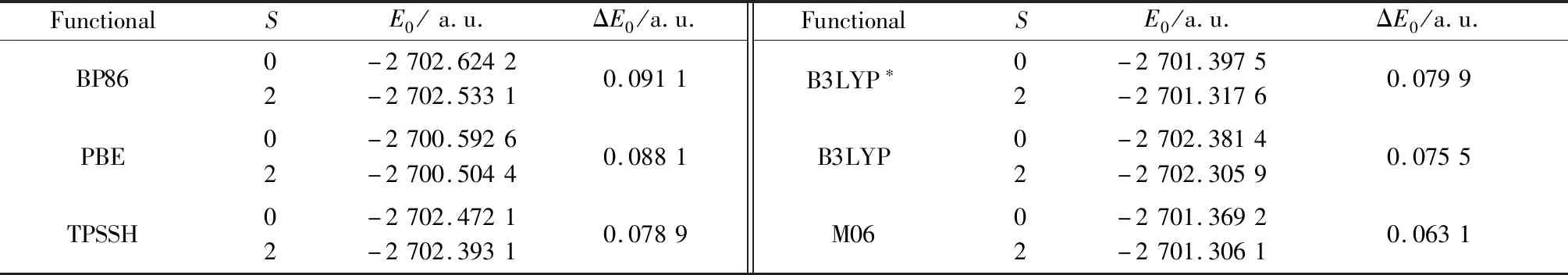
表5 配合物 [Co(2,2-bipy)2(N3)2]+各自旋态(S)在不同泛函下的单点能E0及高低自旋能级差(ΔE0=EHS-ELS)Table 5 Spin (S),calculated single-point energy (E0)and relative energy (ΔE0=EHS-ELS)of [Co(2,2-bipy)2(N3)2]+ calculated by exchange-correlation functionals
2.2.3 配合物的原子电荷及氧化态分析
在B3LYP/def2-TZVP计算水平下,本文使用multiwfn 3.7软件[20],采用原子偶极矩校正的Hirshfeld布居(ADCH)方法[21],分别计算了配合物1阳离子、叠氮离子和2,2-联吡啶配体的原子电荷,计算结果如表6所示。从表中数据可知,中心离子钴(Ⅲ)的电荷由配位前的3.000 0e下降到0.107 461e,说明配体与钴离子发生了配位作用,有部分电荷从配体的N原子转移到了CoIII中心。作为比较,本文还计算了游离的叠氮负离子和2,2-联吡啶的ADCH电荷,结果如表6所示。从表中数据可以看出,与CoIII离子发生配位作用的N原子,其配位后所携带的原子负电荷数都比配位前有不同程度的减小,这也说明了中心金属离子与配体间发生了配位作用,在形成配合物时,配位原子上的电子向金属中心离子发生转移。
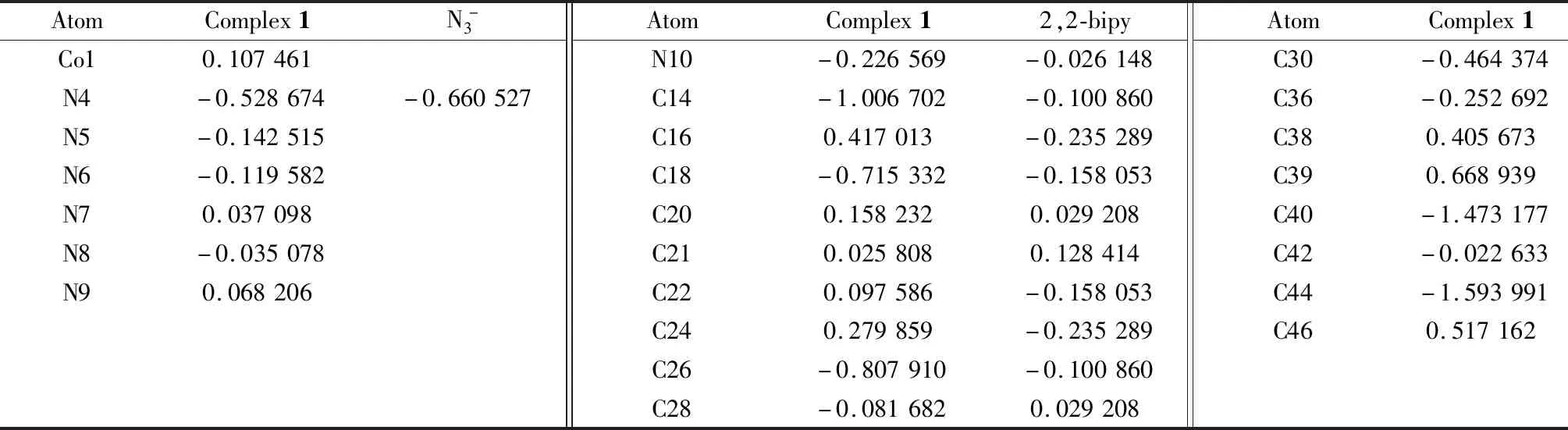
表6 配合物1部分原子、叠氮离子和2,2-联吡啶配体的原子偶极矩校正的Hirshfeld布居(ADCH)原子电荷Table 6 Atomic charge of atomic dipole moment corrected Hirshfeld population (ADCH)for complex 1 anion,N3- and 2,2-bipy /e
此外,本文采用LOBA(localized orbital bonding analysis)方法[22],使用Multiwfn 3.7软件[20]计算了化合物的氧化态,结果显示中心金属Co离子呈+3价,与实验结果相吻合。
2.2.4 前线分子轨道
根据分子轨道理论,最高占据轨道(HOMO)和最低空轨道(LUMO)及邻近轨道对化合物的反应活性影响较大。HOMO及其邻近占据轨道具有优先提供电子的作用,而LUMO及邻近空轨道具有接受电子的作用,HOMO-LUMO轨道能隙值的大小反映了电子从占据轨道向空轨道跃迁的能力,所以一定程度上代表了分子参与化学反应能力的强弱。较大的能隙值意味着较高的稳定性和较低的反应活性。本文使用B3LYP/def2-TZVP方法计算了[Co(2,2-bipy)2(N3)2]+的最高占据轨道和最低空轨道的能量(EHOMO,ELUMO)及其轨道能隙(ΔE)。结果表明,其HOMO和LUMO轨道能量分别为-8.617 8 eV和-5.679 8 eV,能隙为2.938 eV,说明该配合物具有较高的动力学稳定性。图4为标题配合物阳离子的前线分子轨道图,红色和蓝色分别表示正负区域。从图中可以看出,配合物分子的最高占据轨道HOMO,其电子密度主要集中在叠氮负离子上,而最低空轨道的电子密度主要分布于两个2,2-联吡啶配体的吡啶环上。
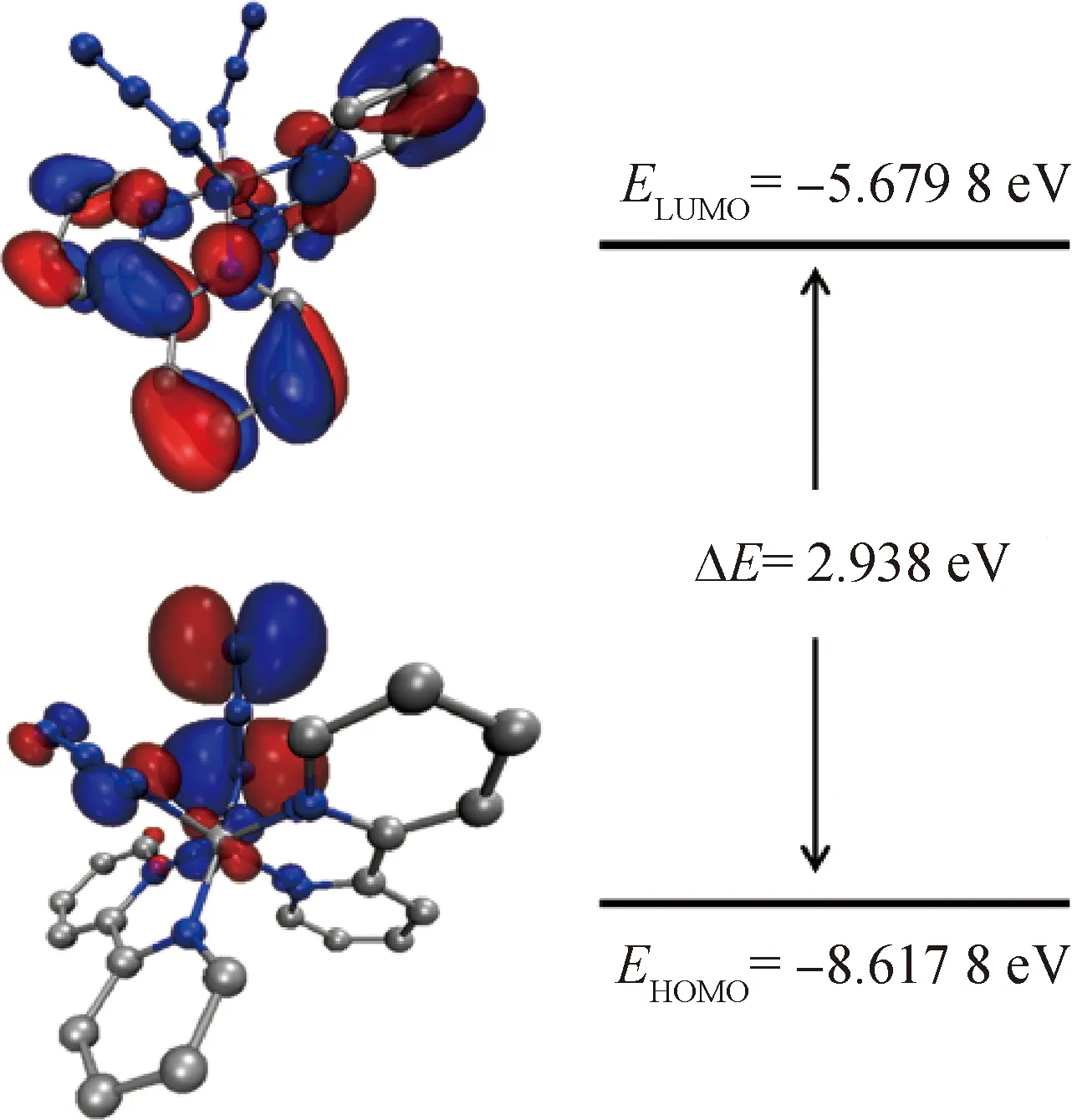
图4 配合物[Co(2,2-bipy)2(N3)2]+的HOMO和LUMO前线轨道示意图Fig.4 HOMO and LUMO of complex [Co(2,2-bipy)2(N3)2]+
3 结 论
本文采用溶剂挥发法合成了一个含有阴离子水簇的带状超分子配合物,并对其进行了结构表征。X射线单晶衍射解析表明,配合物1为含有结晶水的六配位八面体构型的单核CoⅢ配合物,该单核配合物通过分子间C—H…π和π…π相互作用,彼此连接形成带状一维超分子结构。客体水分子与无序的N3-和Cl-通过氢键作用形成了一个包含环状水四聚体的[(H2O)4(N3)Cl]2-阴离子水簇。基于密度泛函理论,对配合物的结构进行了全构型优化和能量计算,分析了其单点能和原子电荷,并计算了中心金属离子的氧化态,计算结果显示,配合物中心钴离子为正三价,低自旋态(S=0),与实验结果相吻合。实验和理论计算分析表明,配合物具有良好的化学稳定性。
