有机合成中化学反应的机器学习
2022-01-09张良顺
张良顺
(华东理工大学材料科学与工程学院,上海市先进聚合物材料重点实验室,上海 200237)
化学反应预测及合成路线设计是有机材料制备的关键步骤之一,然而,高精确度、高效率地预测有机化合物及其逆合成分析仍是极具挑战性的问题,如功能基团的保护、化合物组成片段的遴选等。当前,化学反应预测与合成路线设计的方法可以归纳为3 类:基于规则的专家系统、量子力学模拟以及基于机器学习的系统。基于规则的专家系统可以快速、准确地预测相关性质[1],但该系统也存在一些局限,如需要熟悉化学知识的专家、数据难以成倍扩增、难以预报多步新的化学反应等。基于第一性原理的量子力学模拟可以得到精确的预测结果[2],但是该方法的结果与模型、计算参数等紧密相关;此外,该方法计算量巨大,不易于化学反应和逆合成的高通量预测。作为一种新兴的方法,机器学习可以快速地、并发地预测有机小分子的物理化学性质[3-5],包括化学反应、自由能、能垒等。但也存在着诸多亟待解决的问题,例如数据集不够大和完备、数据偏倚、模型仍缺乏物理基础、获得的结果还不能深入地被阐释等。
针对有机小分子的化学合成,本文综述了机器学习方法在这一领域的进展,包括化学反应数据的收集、化学反应的预测与合成路线的设计等。对于高度复杂的树脂分子,本文论述了基于机器学习合成路线亟待解决的问题。
1 化学反应数据的收集
目前,化学反应数据主要从科学文献、网页和专利中收集[6]。其中,化学专利的文本组织有一定的规律,即首先以合成化合物作为章节的题目,然后是合成步骤的描述,最后是化合物的表征;当化合物合成包括中间物时,每步合成的文本也是单独章节或段落。这些特征有利于化学反应数据条目的文本挖掘。下面以美国专利局的公开专利(即标识为C07 和C08 的国际专利)为例,简要说明化学反应数据条目的文本挖掘流程。当含有标识C07 和C08 专利文本读入以后,判定是否含有多个步骤来区分化学实验文本和非化学实验文本,并进行语义分析(如图1 所示)。具体步骤如下:(1)由专利文本XML 的标识符“heading”或“p”,判定段落的标题。(2)使用朴素贝叶斯分类器判断段落是否为化学实验段落。(3)采用 ChemicalTagger 工具包对标题和段落进行标记,把文本归为化学体和普通短语或词两类。(4)对上面两类分别进行语法分析。化学体的语法分析包括三个方面:结合OSCAR 和OPSIN 工具包,从化学名变换到化学结构,并用SIMILES 和InChI 表达式表示;当化学体为体积、物质的量之比、质量、浓度、pH、产率和物质状态等时,标识为化学性质;分析化学体的属性,包括反应物、产物、溶剂或者催化剂等。普通短语或词的语法分析主要用于识别合成步骤(例如dry,precipitate 和purify 等单词)。(5)化学反应映射。将化学结构的SIMILES 表达式加载到Indigo 工具包,得到化学反应的原子-原子映射。检查产物的原子是否全部来自反应物,以此检验化学反应是否成立。通过化学计量学计算,进一步完善化学反应。(6)化学反应输出。采用图形化和化学标记语言(CML 格式)输出化学反应。其中,CML 格式包含较完整的信息,即反应物和产物及其SIMILES 表达式、性质和状态等。

图1 化学反应数据收录流程图Fig.1 Schematic of the dataset collection of chemical reaction
通过上述步骤,可以收集到格式化的化学反应条目。Lowe 等[7]通过对近30 年美国专利的文本挖掘,构建了开源的USPTO 化学反应数据集。另外,也存在一些商业的化学反应数据集[8]。比如,Elsevier 公司对化学文献进行提取,构建了数据条目更多的Reaxys 化学数据与文献数据库,包括化学结构、特性和反应等。
在获取了化学反应数据集后,可对数据条目的信息进行归纳和总结,例如构建具有精确检索和模糊检索功能的数据库,提取基团间反应模板。其中,应用基于编辑的算法,识别化学反应中参与反应的核心原子,提取基团间反应的特征,建立基团间化学反应的模式,并以SMARTS 规范进行记录; 对基于同种模式的化学反应进行归纳,建立基团间化学反应模板数据库。这些数据条目将在机器学习模型中应用。
2 化学反应的预测
对于给定的反应物、试剂、溶剂等,利用机器学习方法预测或预报其相对应的产物和产物分布,称之为化学反应预测或预报。早期的机器学习研究源于专家推荐反应机理,或者只针对某种特定的化学反应。这类化学反应预报模型在结构上等价于机器学习的回归模型。
随着化学反应数据条目的丰富,并结合近来发展的深度学习方法,科研工作者发展了基于化学反应模板的产物预测方法。例如,Wei 等[9]首先在概念上证实了深度学习预测反应产物的可行性。对于给定的反应物和试剂,利用模拟数据生成与之相近的16 种化学反应模板,从而推演出相对应的产物。Segler 等[10,11]采用实验数据并推广了这种方法,利用算法生成的近万种模板推演出产物的可能性分布;接着,对产物进行评价,评价分数最高的化合物被推荐为主产物。需要指出的是,化学模板推演的化合物可能存在多种非等价的产物,如卤代反应可能存在多个位置选择。
Coley 等[12]应用前向反应模板生成一系列可能发生的反应类型和产物,然后使用机器学习方法评估候选产物中的主反应和产物(如图2 所示)。具体步骤为:(1)前向反应枚举。对于每个原子映射反应的SMILES 表达式,反应核定义为反应物原子的连接发生变化的反应。通过邻近非映射原子和键的重排,此反应核可以衍生其他可能的反应,并用SMARTS 表达式表示。这些衍生反应组成可能的化学反应,产生一系列候选产物。(2)候选产物排序。编辑基反应表述用于描述反应核中原子连接的改变。一个候选的原子映射反应可以解析成4 种编辑类型:原子ɑi失氢;原子ɑi加氢;2 个原子ɑi和ɑj成键;2 个原子ɑi和ɑj断键。前两者含有反应物原子的32 个特征,而后两者有68 个特征。基于反应可能性与原子或键改变的有关事实,设计神经网络。首先,对于单个编辑类型,建立全连接的神经网络,使编辑基反应表述转变到矢量基表述。然后,全部编辑类型的矢量进行加和,传递到下一个神经网络,从而计算出一个标量值。对于一个候选反应,此标量值表示该反应的发生倾向。通过softmax 层,全部候选反应的标量值变换成发生反应的可能性。最大值为最可能发生的反应,对应的产物为主产物。该方法能够预测大多数产物,但预测准确率依赖于模板质量以及训练数据量和特征等。

图2 化学反应预测模型的示意图(插图为全连接神经网络)[12]Fig.2 Schematic of machine-learning model for the prediction of chemical reaction(Inset shows the fully connected network)[12]
虽然上述基于模板的反应预测模型能较高精度地预测主产物,但产物局限于已知模板的预测范围。这限制了机器学习模型预测或预报新产物的可能性。为了克服此局限,无模板的化学反应预测模型被提出。一类是针对化合物SMILES 表达式的Sequence-to-Sequence(Seq2sep)模型[13,14]。这一模型的思路来自化学反应与机器翻译之间的可类比。也就是,化学反应对应于反应物和试剂的SIMILES 字符串转换成不同长度的产物SIMILES 字符串。借鉴机器翻译的Seq2sep 模型,并融合注意力机制,实现了无模板的化学反应预测。在此基础上,通过SIMILES 表达式的语法校正等改进方案,可进一步提升化学反应预测的精确度。需要提及的是,Seq2sep 模型的训练需要极大的化学反应数据集。
利用图卷积神经网络,Coley 等[15]提出了另外一类无模板的化学反应预测机器学习模型,如图3 所示。不同于SIMILES 表达式的格式化输入,化合物分子以非格式化图表示,特征包括结构信息(如原子数、质量、芳香性、连接性、键态等)以及容易计算的一些几何和电子信息(如局部电荷、疏水性表面积等)。通过Weisfeller-Lehman 网络,化合物分子中原子信息嵌入到神经网络中。而反应物和试剂关系通过全局注意力机制表达。以此作为全连接网络的输入,预测原子间的活性或化学反应中心。最高活性的原子被用于枚举可能的反应产物。通过结构和价态的有效判定后,用Weisfeller-Lehman 差分网络评价有效产物的分数。最高者为最可能的化学反应产物。相比于其他方法,图卷积神经网络具有可物理解释性。但是,该方法预测产物的精确度非常强地依赖于数据集的选择。解决方案之一是进一步丰富非格式化的图表示[16]。
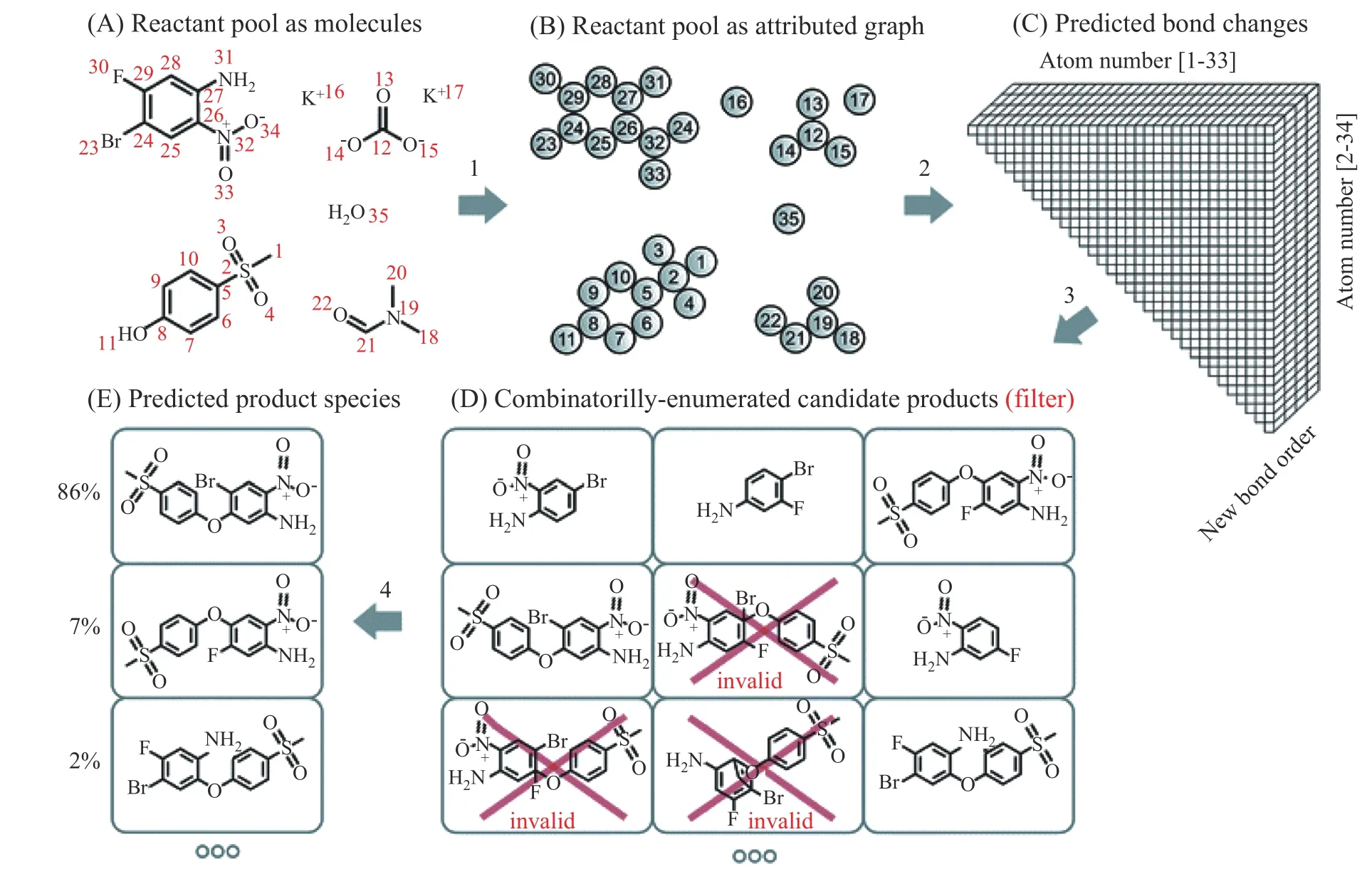
图3 化学反应预测的图卷积神经网络模型[15]Fig.3 Modelling of graph-convolution nerve networks for the prediction of chemical reaction[15]
产物的产率不仅与反应物和试剂有关,还依赖于反应条件(计量比、浓度、温度和时间等)。从数学角度,产率的提升对应于反应条件的优化问题[17-19]。机器学习模型也适用于化学反应条件的推荐。最有可能与实验操作相结合的机器学习方法是主动学习方法。对于反应条件,构建能代表产率的代理机器学习模型,采用Bayesian 优化[20],寻找下一个反应条件,优化模型直至达到最优反应条件。
目前,化学反应产物的预测还存在一些亟待解决的问题:其一,化学反应数据集包含产物、反应物和试剂等信息,但与产物预测相关的催化剂、溶剂和温度等变量的信息不完备。同时,产物通常是产率超过50%的主产物,而副产物的信息也不完备。其二,在机器学习模型中产物的评价分数与合成条件、热力学态和活化能等无关联。这阻碍了可合成、热力学稳定化合物的遴选以及高效合成。其三,化学反应数据条目非常多,导致化学反应预测机器学习模型的计算量巨大。可能解决方案是利用特定的化学反应作为模型训练的数据子集,同时包含更多的特征量(如表面电荷、偶极矩以及活化能等),从而在增加较小的计算量时提升特定化学反应的预测精度和效率。
3 合成路线的设计
对于给定的有机化合物分子,寻找合成目标分子的可能反应物,以可能反应物分子为次级目标分子,重复上述过程,直至反应物为易获取分子。这一过程称之为化合物的合成路线设计。目前,机器学习方法已经应用于合成路线的设计。依据化合物生成规则,合成路线的机器学习方法可分为基于模板和无模板两类。与基于模板的化学反应预测方法相似,基于模板的合成路线设计需要非常深厚的专业知识,产物逆合成局限于已知模板的合成路线设计范围。而无模板的机器学习方法能克服上述障碍。下面以无模板合成路线设计为例,阐述机器学习在此领域的拓展。
Liu 等[21]采用Seq2seq 模型预测有机小分子的单步逆合成,如图4 所示。具体步骤包括:(1)Seq2sep 模型生成反应物。Seq2seq 模型可以从一个序列映射到另一个不等长度序列。模型包括编码(Encoder)和解码(Decoder)部分,每部分含有循环神经网络。Encoder 部分完成产物分子的编码工作,将不同的输入编码转换成一个定长的向量;Decoder 部分则完成反应物解码工作,对编码器的结果进行解码输出,即反应物。(2)评估和预测。解码的输出为预测的反应物以及log 似然函数值。当log 似然函数值最小时,则认为是最可能的反应物。该模型的预测精度约为65%(Top-5)。通过语法校正,可进一步提升预测精度[22]。

图4 逆合成反应预测示意图(插图为Seq2seq 模型)[21]Fig.4 Schematic of machine-learning model for the retrosynthesis(Inset shows the Seq2seq model)[21]
以上工作只对产物进行了单步逆合成研究。对单步逆合成的多步递归,并对反应物进行树形检索,可得到产物的合成路线[23]。Schwaller 等[24]组合分子的Seq2seq 模型和图搜索策略,实现了无人工干预的合成路线设计。Shibukawa 等[25]利用深度优先搜索算法构建化学反应网络,以此枚举全部可能的合成路线,利用评价分数筛选可能的合成路线,并以Cetirizine 药物分子为例证实该思路有效。
需要指出的是,反应物的评价方式有许多[26]。例如,依据SIMILES 文本序列长度,SA_Score 测度可定义为原子数、成环原子数以及手性原子数的函数,但是在实验中产物合成还与反应物、试剂的可获取性和脱保护等紧密相关。SCScore 测度考虑到上述真实合成情况。以Reaxy 提取的反应为训练数据集,神经网络可用于计算合成复杂度SCScore。通过比较不同的评价方式,证实SCScore 测度对合成复杂性的描述较优异。
4 树脂分子有机合成的思考
不同于目前研究比较多的简单分子或天然化合物,树脂分子的特点有:三维网络状、拓扑定义不明确、杂环且高分子量等。其材料性质和功能也与其合成策略紧密相关。目前,机器学习用于树脂分子的有机合成、分子设计-性质关系的研究仍处于探索期。针对树脂分子的内在特点,以机器学习视角思考有机合成中亟待解决的问题和可能的方向。
4.1 缺乏丰富的数据
目前,USPTO、Pistochio、Reaxy 等化学反应数据库包含少量的高分子和极少的树脂分子的数据条目。原因之一是高分子量化合物的自动合成技术不成熟。另外一个原因是该类化合物分子结构与合成工艺、催化剂等相关。可行的方案是人工干预的数据收集。
4.2 无规范化的表示
存在多种格式表示聚合物分子的结构和拓扑,但适用范围有限。Pistoia 联盟的HELM 格式只适用于明确定义的大分子。国际纯粹与应用化学联合会的InChI 格式不支持支化聚合物。CurlySMILES 格式可适用于复杂高分子,但是语法过于复杂。BigSMILES 格式可支持复杂拓扑的聚合物体系[27],但其应用还有待进一步验证。
4.3 基于图网络的有机合成模型
有机分子非常适合于以无结构化的图表示。图卷积网络模型也已经应用于化学反应的预测和合成路线的设计,但是,当训练数据非常局限或偏倚时,图卷积网络模型的预测准确率并没有优势。对于树脂分子的有机合成、分子设计-性质关系的研究,在少样本情况下实现高准确率和探索与开发是图网络模型亟待解决的问题。
4.4 主动学习的运用
机器学习的预测结果需要模拟、实验等其他方法验证,或反馈到机器学习模型中[28]。对于无明确拓扑的树脂分子体系,模拟和实验的开展需要长时间、合成技巧等。利用主动学习方法自主地选择最可能的物理化学参数、工艺参数等,将加速有机合成策略或分子设计的优化。
4.5 基于人工智能的合成平台
结合树脂分子的合成路线规划、流动合成系统、机械手辅助的批次合成系统等,通过简单易得的原材料,经过化学反应,在无人工干预、自主判定最优工艺条件下获得拓扑定义明确但结构复杂的高分子量化合物[29,30]。
5 总结
本文综述了机器学习方法在有机小分子化学合成领域的研究进展,包括化学反应数据的收集、化学反应的预测和合成路线的设计等3 个方面。对于三维网络状、拓扑定义不明确的树脂分子,论述了基于机器学习合成路线的亟待解决问题,例如缺乏丰富的数据、无规范化的表示、少样本的机器学习模型等。
