基于16S rRNA 高通量测序技术的肠道菌群多样性推断死亡时间
2022-01-07曹洁李文晋王一飞安国帅卢晓军杜秋香李晋孙俊红
曹洁,李文晋,王一飞,安国帅,卢晓军,2,杜秋香,李晋,孙俊红
1.山西医科大学法医学院,山西 太原 030001;2.包头市公安局刑事侦查支队,内蒙古 包头 014030;3.山西医科大学第二医院,山西 太原 030001
死亡时间又称死后间隔时间(postmortem interval,PMI),通常指死亡发生到尸体被发现的间隔时间[1]。死亡时间推断是法医学鉴定中需要解决的重要问题之一,对确定案发时间、认定和排除嫌疑人、划定侦查范围具有重要意义。传统的PMI 推断主要依据死后尸体现象变化规律粗略判断。随着分子生物学技术的发展,实时荧光定量聚合酶链反应(real-time fluorescent quantitative polymerase chain reaction,qPCR)技术[2]、双向电泳(two-dime nsional electrophoresis,2-DE)和高效液相色谱-质谱(highperformance liquid chromatography-mass spectrometry,HPLC-MS)技术[3]、傅里叶变换衰减全反射红外光谱(attenuated total reflectance-Fourier transform infrared,ATR-FTIR)技术[4]等越来越多的新型技术被应用到PMI 推断研究中。大量相关研究为寻找便捷快速的技术方法、敏感的指标以及建立合适的数学模型以缩小PMI 窗口提供了实验基础和新的思路。
微生物在周围环境和个体中无处不在,生态环境中的微生物在地球化学循环中发挥着重要的生态作用,人体内的微生物更是广泛参与免疫、消化、代谢等生理过程[5]。肠道微生物在食物分解、药物降解、脂肪积累、维生素和氨基酸合成中扮演着重要的角色[6]。肠道微生物不仅参与体内的稳态代谢,而且影响尸体分解的发展和进程,在尸体腐烂过程中发挥着重要作用[7]。机体死亡后,微生物既帮助分解机体组织,又通过分解代谢完成物质的自然循环[5]。
既往对肠道微生物的研究主要基于培养法,而能够培养的微生物仅占微生物种类总数的1%[8]。随着高通量测序技术的成熟与发展,细菌群落结构分析被广泛应用。不同环境中的微生物多样性被逐一解读,大样本、高通量的测序可对微生物变化进行动态的、系统的分析。有研究[9]表明,尸体降解过程中肠道厚壁菌门(Firmicutes)和拟杆菌门(Bacteroidetes)的数量减少,而变形菌门(Proteobacteria)的数量增加,提示肠道微生物群落变化具有较强的时间变化规律,而肠道微生物在尸体腐败破坏之前,处于相对封闭的环境,其菌群的变化主要与PMI 相关,有望成为精确推断PMI 的有效方法。
本研究收集了大鼠死后30 d 内不同时间点盲肠内容物,采用16S rRNA 高通量测序技术探究大鼠肠道菌群与PMI 之间的关系,从多方面分析比较各时间点间差异,建立偏最小二乘(partial least squares,PLS)回归模型,寻找时序性变化特征,从而更准确地推断PMI。
1 材料与方法
1.1 大鼠死亡模型制备及检材提取
健康成年雄性Sprague-Dawley大鼠84只,10~12周龄,体质量250~300 g,由山西医科大学实验动物中心提供。温室饲养2 d 后,采用断颈方式处死大鼠,死后存放于16 ℃人工气候箱。于死后0、1、2、3、5、7、9、12、15、18、21、24、27 和30 d(每个时间点6 只大鼠)共14 个时间点分别取大鼠盲肠内容物置于干净的微量离心管内,立即放入液氮中冷冻,随即将样本保存至-80 ℃冰箱待检。
本实验由山西医科大学科学研究伦理审查委员会批准(审批号2018LL351)。
1.2 DNA 提取及浓度、纯度检测
取200 mg 盲肠内容物于新微量离心管中,使用QIAamp PowerFecal Pro DNA 试剂盒(德国Qiagen 公司)提取总DNA,具体操作步骤参照试剂盒说明书。使用Infinite®200 PRO 酶标仪(瑞士TECAN 公司)检测总DNA 纯度及浓度,光密度D260/D280在1.8~2.0 的DNA 样本用于后续实验。
1.3 文库构建及高通量测序
DNA 质检达要求后,以此为模板,采用带barcode的引物对16S rRNA 的V3~V4区进行第一轮PCR 扩增,扩增引物为341F(5'-CCTACGGGNGGCWGCAG-3')和805R(5'-GACTACHVGGGTATCTAATCC-3'),反应体系30 μL:15 μL 即用型的常规PCR 预混合溶液(上海翊圣生物科技有限公司),上下游引物各1 μL,10~20 ng DNA 模板,9~12 μL ddH2O。随后引入二代测序接头引物试剂盒(Illumina 平台Index Primer13-24,北京百奥莱博科技有限公司)进行第二轮PCR 扩增,反应体系30 μL。用20 g/L 琼脂糖凝胶电泳检测文库大小,检测合格后,为了得到均匀的长簇效果和高质量的测序数据,使用Qubit®3.0 荧光定量仪(美国Thermo Fisher Scientific 公司)进行文库浓度测定。通过Illumina Miseq PE300 测序平台(美国Illumina 公司)对DNA 片段进行双端高通量测序,获得原始数据。
1.4 数据预处理
得到原始数据后,用Cutadapt v1.2.1(https://cutadapt.readthedocs.io/en/stable/)软件去除接头序列,将成对的短序列用PEAR v0.9.6(https://cme.h-its.org/exelixis/web/software/pear/)软件依据重叠关系进行数据拼接。采用PRINSEQ v0.20.4(http://prinseq.sourceforge.net/)软件切除barcode 序列和引物序列,并采用滑窗法检查各个序列的质量值,以去除含N 部分序列、短序列以及低复杂度序列,长度阈值为200 bp。使用USEARCH v8.1.1831 软件(www.drive5.com/usearch)去除预处理后序列中非扩增区域序列,并用UCHIME v4.2.40 软件(http://drive5.com/usearch/manual/uchime_algo.html)检测嵌合体并予以删除,随后去除嵌合体的序列与核糖体数据库项目(Ribosomal Database Project,RDP;http://rdp.cme.msu.edu/misc/resources.jsp)代表性序列进行比对,剔除相似度低于80%的序列,得到合格序列。USEARCH v8.1.1831软件将上述数据进行97% 相似性运算分类单元(operational taxonomic unit,OTU)聚 类,采用RDP Classifier v2.12 软件(https://sourceforge.net/projects/rdp-classifier/files/)将各样本序列进行物种分类,选择丰度最高的序列作为OUT 代表性序列。
1.5 统计分析
在门、属水平对微生物群落构成及分布丰度进行分析。用R v3.2(https://www.r-project.org/)对物种分类学统计结果做柱状图,云工具(https://www.lc-bio.cn,杭州联川生物技术股份有限公司)绘制桑基图观察微生物群落构成。基于前述OTU 聚类结果进行α多样性分析[10],使用Mothur v1.30.1 软件(https://mothur.org/)计算各个样本α多样性指数值(ACE 指数和Shannon 指数),检验水准α=0.05。线性判别分析效应大小(linear discriminant analysis effect size,LEfSe)是一种线性判别分析(linear discriminant analysis,LDA)方法[11],结合Wilcoxon 和Kruskal-Wallis 非参数检验可以筛选出最能解释组间差异的微生物菌群,以及其对组间差异的影响程度,本研究使用LEfSe v1.1.0 软件(http://huttenhower.sph.harvard.edu/lefse/)选择各时间点LDA 分值>2 的差异细菌群落。利用SIMCA®14.1(Q845)软件(德国Sartorius 公司)建立PLS 回归模型[12],得到决定系数(R2)和交叉验证均方根误差,分析PMI 与肠道菌群之间的线性关系。
2 结果
2.1 样本测序结果
本研究对大鼠死后14 个时间点共84 个样本进行测序,获得原始数据8 282 115 条,经数据预处理获得有效数据为6 591 968 条,每条序列平均长度为(416.11±4.09)bp。根据相似性将上述序列聚集成为23 759 个OTU,采用RDP Classifier v2.12 软件对OTU代表序列进行物种分类,共得到28个门、53个纲、87个目、163个科、347个属,用于后续不同水平的菌落组成分析。
2.2 门、属水平微生物群落构成及分布丰度分析
在门、属水平分别选择丰度最高的前20 个(>1%)物种做柱状图进行分析(图1A~B)。门水平(图1A),厚壁菌门在0 d 平均相对丰度最高,达到近80%,随后整体呈下降趋势,但在各时间点相对丰度始终最高;拟杆菌门相对丰度在死后0~7 d 上升,9 d 后下降,变形菌门在死后12、18、21 d 相对丰度高于其他时间点。厚壁菌门、拟杆菌门、变形菌门在整个样本中的相对丰度最高,其余前10 大菌门分别为放线菌门(Actinobacteria)、疣微菌门(Verrucomicrobia)、糖念珠菌门(Candidatus Saccharibacteria)、螺旋体门(Spirochaetes)、广古菌门(Euryarchaeota)、浮霉菌门(Planctomycetes)、酸杆菌门(Acidobacteria),在分解过程中所有时间点相对丰度都比较低。属水平(图1B),乳酸菌(Lactobacillus)在0、3、5、7、9、12、15、21、24、27、30 d 相对丰度最高,其余前10 大菌属分别为罗姆布茨菌属(Romboutsia)、肠球菌属(E.enterococcus)、拟杆菌(Bacteroides)、双歧杆菌(Bifidobacterium)、巴内氏菌属(Barnesiella)、阿克曼氏菌属(Akkermansia)、梭状芽孢杆菌XlVa(ClostridiumXlVa)、梭状芽孢杆菌(Clostridium sensu stricto)、假单胞菌(Pseudomonas),其相对丰度都比较低。
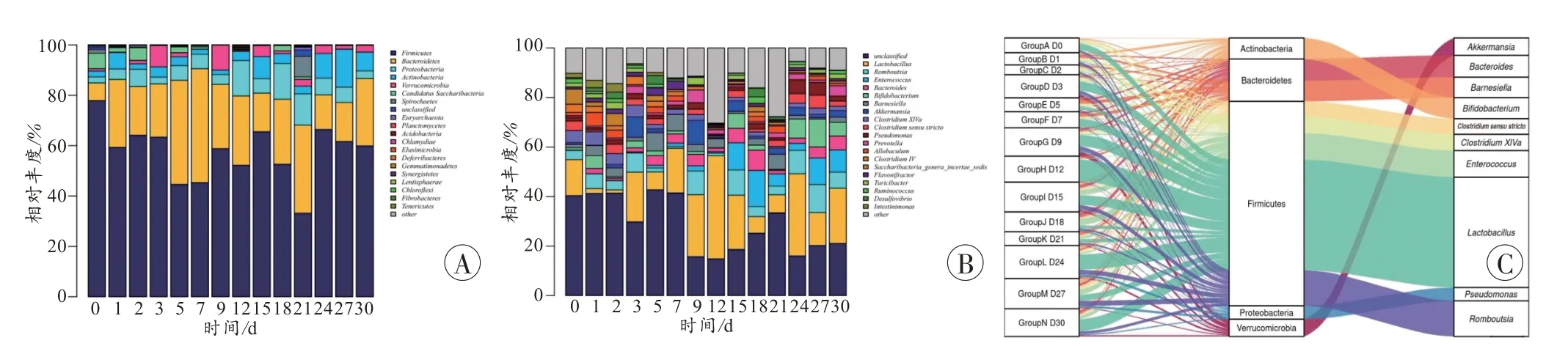
图1 不同水平微生物群落相对丰度Fig.1 Relative abundance of microbial communities at different levels
桑基图(图1C)中,不同颜色彩色条带代表不同菌属,可以直观地看到相对丰度前10 的菌属都属于相对丰度前5 的菌门,左边与各时间点相连的条带端点宽度表示菌属在该样本中的丰度,不仅反映了每个时间点的优势菌群组成比例,同时也反映了各优势菌群在不同时间点之间的分布比例。
2.3 α 多样性分析
由表1 可知,除死后5 天微生物群落总数升高(P<0.05)外,其他PMI 点在数量上差异无统计学意义;与0 d 相比,死后9、12、15、24、27、30 d 的Shannon指数降低(P<0.05)。

表1 肠道微生物菌群α 多样性分析Tab.1 Alpha diversity analysis of intestinal microbial communities(n=6,x¯ ±s)
2.4 细菌群落结构差异统计分析
图2 为LDA 分值分布柱状图,对14 个时间点的细菌群落进行比较,在13 个时间点(除第24 天)共筛选出119 个具有差异的细菌群落。

图2 线性判别分析效应Fig.2 LDA effect
2.5 PLS 回归模型建立
根据全部时间点实际值与预测值建立的PLS 模型(图3A),其R2为0.795,交叉验证均方根误差为6.57 d。根据9 d 前样本和12 d 后样本分别建立PLS回归模型(图3B~C),两者R2、交叉验证均方根误差分别为0.767、1.96 d 和0.445、5.37 d。

图3 PLS 回归分析Fig.3 PLS regression analysis
3 讨论
随着16S rRNA 高通量测序技术的成熟与发展,肠道菌群的研究在疾病诊断、临床治疗等[11,13-14]方面均有应用。近年来,有学者将死后微生物的时序性变化用于PMI 推断的研究。METCALF 等[5,15]研究了小鼠和人尸体皮肤、腹腔、周围土壤在不同季节下微生物随PMI 变化的关系,建立回归模型推断PMI。LIU 等[16]评估了死后15 d 内微生物群落结构,基于随机森林(random forest,RF)、支持向量机(support vector machine,SVM)、人工神经网络(artificial neural network,ANN)等算法建立模型推断PMI。本研究通过分析大鼠死后30 d 内不同时间点肠道菌群测序结果,探索其与PMI 之间的关系。
桑基图是样本与物种的共现性关系图,描述样本与物种间对应关系的可视化图,从图中可见相对丰度最高的前10 个菌属都属于丰度最高的前5 个菌门,反映每个样本的优势物种组成比例,同时也反映各优势物种在不同样本之间的分布比例。本研究结果显示,在门水平厚壁菌门、拟杆菌门、变形菌门相对丰度最高,且各时间点α多样性指数表明,随时间推移,微生物群落总数未发生明显变化,而菌群多样性却在多个死后时间点明显升高(图2)。上述针对各采样时间点微生物群落在门、属水平发生的变化,与LI 等[17]的研究结果基本一致,但本研究探索了死后30 d 内更多时间点的肠道菌群变化规律。
LEfSe 分析可得到LDA 分值分布柱状图和组间差异具有统计学意义的差异物种在不同组别的丰度。本研究发现,在14 个时间点中除第24 天外其余13 个时间点共筛选出119 个具有差异的细菌群落,第24 天没有差异的细菌群落的原因可能为随着时间的推移,菌群多样性下降,没有LDA 分值>2 的差异菌群。
如何通过合理的建模方式提高PMI 推断的准确性是一个亟待解决的问题,PLS 回归是目前最常用的建模方法[12,18-19]。为明确OTU 水平与PMI 的相关性,本研究建立了PLS 回归的PMI 推断模型,结果显示,R2为0.795,但交叉验证均方根误差为6.57 d。若将间隔时间较短的9 d 前和间隔时间较长的12 d 后分别做PLS 回归,其R2、交叉验证均方根误差分别为0.767、1.96 d 和0.445、5.37 d,说明通过分段建模的方式可以分别提高9 d 前和12 d 后的模型推断准确性,提示在PMI 推断中应避免采用单一统计方法进行PMI 推断,运用多段建模的方式可提高其准确性。
肠道菌群多样性容易受个体饮食习惯、临时性用药及周围环境变化、死后腐败等多种因素的影响[7],如何有效避免上述因素对与PMI 具有高度相关的菌群变化的影响是肠道菌群用于PMI 推断的关键。本研究在实验设计中并未考虑动物模型的致死方式(如麻醉药)及周围环境对肠道菌群多样性的影响,因此在今后的研究工作中应考虑尸体致死方式、周围环境(温度、湿度等)、土壤类型、土壤质地、昆虫等[9,20-21]影响因素,同时继续延长死后时间,增加时间点,提高实验数据准确度,并尽可能减少误差。
综上所述,利用16S rRNA 高通量测序技术检测的肠道微生物在死亡30 d 内呈现一定时序性变化,采用分段建模的方式可以提高死亡9 d内和死后12~30 d的准确性。
