The gut microbiome and metabolome of Himalayan Griffons (Gyps himalayensis):insights into the adaptation to carrion-feeding habits in avian scavengers
2022-01-07WenWangXiaolongGaoSisiZhengZhuomaLancuoYingLiLilinZhuJianpingHouJiayiHaiXinLongHanxiChenAlexeyDruzyakaandKirillSharshov
Wen Wang , Xiaolong Gao, Sisi Zheng, Zhuoma Lancuo, Ying Li, Lilin Zhu, Jianping Hou, Jiayi Hai,Xin Long, Hanxi Chen, Alexey Druzyaka and Kirill Sharshov
Abstract
Keywords: Avian scavengers, Culture, Microbiome, Pathogens, Vultures
Background
Vultures, known as “nature’s clean-up crew”, are large raptors that feed predominately on the animal carcasses without causing any adverse effects. For millennia, the special scavenging activities of vultures benefit the health of both human and wildlife populations by reducing the transmission of pathogens resident in rotting carcasses across a wide variety of landscapes and habitats(Moleón et al. 2014). There are 23 extant vulture species in the world and composed of two independently evolved clades, including the New World vultures (7 species in the avian family Cathartidae) and Old World vultures (16 species in the avian family Accipitridae) (Jarvis et al. 2014). About 81% of the 16 Old World vulture species are threatened or near-threatened at the global level and many vulture species exhibit regional contractions in both abundance and distribution (Ogada et al.2016; Margalida and Oliva-Vidal 2017). Among those,the Himalayan Griffon (Gyps himalayensis
) is the most widely distributed and abundant vulture species concentrated on the Qinghai-Tibetan Plateau, where livestock carcasses (most of them are yaks and Tibetan sheep raised by local herdsmen) are left for Himalayan Griffons for consumption. Due to the rapid decline in Himalayan Griffon populations in the last few decades, the status of this species in the International Union for Conservation of Nature’s (IUCN) Red List was changed from “Least Concern” in year 2012 to “Near Threatened” in year 2014(BirdLife International 2017). Himalayan Griffons are now a second-class national-protected bird in China and are legally protected. As the resident and relatively sedentary vulture species on the Qinghai-Tibetan Plateau,the Himalayan Griffons are crucial to maintain the balance of the Plateau ecosystem and the Tibetan sky-burial culture (Lu et al. 2009).One of the most central aspects of vulture biology is how they protect themselves against the microbial toxins and pathogens in their food sources and scavenging lifestyle. Physiologic, genomic, transcriptomic and metagenomic studies of different species of vultures have revealed a wide variety of adaptive and protective mechanisms of these avian scavengers adapted to the scavenging diets (Mateos-Hernndez et al. 2013; Roggenbuck et al. 2014; Chung et al. 2015; Blumstein et al. 2017;Zepeda Mendoza et al. 2018). For example, vultures were reported to have an especially low gastric pH that could kill pathogenic bacteria ingested from carrion (Houston and Cooper 1975). Analysis of the whole genome of the Cinereous Vulture (Aegypius monachus
) (Chung et al.2015) and the Turkey Vulture (Cathartes aura
) (Zhang et al. 2014; Zhou et al. 2019) identified a series of genes and pathways contributed to strengthen the immune defense against pathogenic invasion. Besides, based on deep metagenomic sequencing, the specialized fecal and gut microbiome was found to be involved in prevention growth of pathogens (Roggenbuck et al. 2014; Zepeda Mendoza et al. 2018), hinting vulture-associated gut microbiome might play an important role in the adaptation of vultures to scavenging.With the considerable progress made in next-generation sequencing techniques, there has been increasing interest in gut microbiome (the collection of gut microbes and their genomes and metabolites) as accumulating studies showed that microorganisms within the gut play an important role in several fundamental and crucial processes such as development, immune homeostasis, nutrient assimilation, vitamins synthesis, and diseases related to humans and other animals (Kau et al.2011; Lee and Hase 2014; Colston and Jackson 2016). The gut microbiome is also important for bird species (Kohl 2012; Waite and Taylor 2014, 2015; Hird 2017), not only aiding digestion and promoting nutrient absorption and energy utilization, but also involved in the behaviors (e.g.mating, recognition, and migration) (Lee 2015), detoxification (Kohl et al. 2016), and immune regulation (Kogut 2017). In addition, analyses of gut microbiomes have been reported to have important applications in wildlife conservation practices (Trevelline et al. 2019; Wei et al.2019). However, even on a global scale, information on the structure and function of gut microbiome related to vulture species is scarce (Kocijan et al. 2009; Roggenbuck et al. 2014; Meng et al. 2017c; Zepeda Mendoza et al. 2018), particularly in the Himalayan Griffons. The only gut microbiome study of Himalayan Griffons to date is published by Meng et al., where the authors firstly sequenced the microbiomes of only three Himalayan Griffons’ rectal swabs and found that Himalayan Griffons were an important reservoir forClostridium perfringens
(Meng et al. 2017c). Different from Meng et al. (2017c),we attempt to study the gut microbiome from the perspective of assisting the Himalayan Griffon host to adapt to scavenging.In order to deepen the knowledge and gain new insights into the Himalayan Griffons’ gut microbiome,we characterized the structure and functions of microbiomes using a combination of culture-independent and dependent methods. The microbiome of different types of carrion and the fecal metabolome were also further explored in this study. The results not only provide new insights into the scavenging adaptation of the Himalayan Griffons but also give information which is crucial for the conservation of this species.
Methods
Ethics statement
This study conformed to the guidelines for the care and use of experimental animals established by the Ministry of Science and Technology of the People’s Republic of China (Approval number: 2006-398). The research protocol was reviewed and approved by the Ethical Committee of Qinghai University. This study did not involve capture or any direct manipulation or disturbance of wild Himalayan Griffons in the fieldwork.
Study site, subjects, and sample collection
A total of 28 fresh fecal samples were collected from 8 different foraging flocks of Himalayan Griffons at 8 locations in Qinghai Province, China (Fig. 1). The coordinates of the 8 sampling flocks, the number of individuals and the feces collected in each foraging flock, and the type of carrion that each flock feeds on were listed in Additional file 1: Table S1. Fecal sample collection in all groups was performed when Himalayan Griffons feed, making sure only fresh feces were being collected. Also, samples were collected from the core of the fecal material, avoiding from the sides touching the ground. Furthermore,the fecal samples were collected at a minimum distance interval of 5 m, and only a small number of fecal samples were collected from each foraging flock to ensure that each fecal sample comes from the different individual of Himalayan Griffon. These samples were provisionally frozen at − 20 °C in a portable freezer during the field work,and subsequently transported to the laboratory, and stored at − 80 °C for microbiome analyses. At the same time, 12 of the fecal samples were divided into two parts,one part for gut microbiome sequencing as described above and the other part for liquid chromatography-mass spectrometry (LC–MS) untargeted metabolomic analyses. An additional set of fecal samples was collected in 1.5 mL Eppendorf tubes, and stored at − 4 °C in a portable freezer, and shipped back to the laboratory for gut bacteria isolation and identification. While collecting fecal samples, we used sterile cotton swabs to collect 8 different carrion’s microorganisms. These sterile cotton swabs were then loaded into 1.5 mL sterile Eppendorf tubes and transported back to the laboratory, and stored at − 80 °C for microbiome analyses.
Fig. 1 A map of sampling sites
High‑throughput Illumina sequencing of the V4–V5 regions of the 16S rRNA genes DNA extraction, PCR amplification, and Illumina MiSeq sequencing
Extraction of microbial DNA from a total of 28 feces and 8 carrion’s swabs was performed using the E.Z.N.A.Bacterial DNA Kit (Omega Bio-tek, Norcross, GA, USA)following the manufacturer’s protocols. The extracted DNA concentration and purification was checked by Qubit 2.0 Fluorometer (Life Technologies, Carlsbad, CA,USA) and 1% agarose gel electrophoresis, respectively.The V4–V5 hyper-variable regions of the bacteria 16S rRNA genes were amplified using the universal primer sequence 515F (5′-GTG CCA GCMGCC GCG GTAA-3′) and 909R (5′-CCC CGY CAA TTC MTTT RAG T-3′), with an Illumina adapter sequence on the forward primer, and an eight-base barcode unique to each sample attached to the 3′ end of the forward primer. The PCR reactions were performed as follows: an initial denaturation at 95 °C for 3 min; first 5 cycles of 30 s at 95 °C for denaturation, 30 s at 45 °C for annealing, 30 s at 72 °C for primer extension, then 20 cycles of 95 °C denaturation for 30 s, 55 °C annealing for 30 s, and 72 °C extension for 30 s, and a final extension of 72 °C for 5 min. The PCR reactions were performed in 20 μL mixture containing 2 × KAPA HiFi HotStart Ready Mix (Kapa Biosystems,Wilmington, MA, USA) 10 μL, 2.5 mM dNTPs, 10 μM of each primer, and 20 ng of template DNA. All reactions for one sample were amplified in duplicate and combined prior to purification. All amplified PCR products were extracted from 2% agarose gel and further purified using the AxyPrep DNA Gel Extraction Kit (Axygen Biosciences, Union City, CA, USA).
Bioinformatic analysis
Raw forward and reverse reads were demultiplexed,quality-filtered, and joined using QIIME (version 1.9.1)(Caporaso et al. 2010), with the following criteria: (1)120-bp reads were truncated at any site receiving an average quality score < 20 over a 50-bp sliding window, and the truncated reads shorter than 50 bp were discarded; (2) reads must be exact barcode matching, with 2 nucleotide mismatch in primers matching,and containing ambiguous characters were removed;(3) only sequences overlapping over more than 10 bp were assembled according to the overlapping sequence.Reads which could not be assembled were discarded.High-quality reads were clustered at 97% sequence identity into operational taxonomic units (OTUs) using the UPARSE pipeline (Edgar 2013). Flower plot was implemented using the R package to show unique and shared OTUs. OTU level core microbiome analysis was carried out using QIIME platform. Increments begin at 50% of the sample number, and then add up to 5%to calculate the number of common OTUs contained in the samples of a certain percentage. The taxonomy of each 16S rRNA gene sequence was analyzed by RDP Classifier (http:// rdp. cme. msu. edu/) against the Silva 16S rRNA database (Release128 http:// www. arb- silva.de) using a confidence threshold of 80% (Quast et al.2013). The species accumulation curve was plotted to reflect the sampling sufficiency. For alpha diversity analysis, species richness (Chao1 index) and evenness (Shannon index) were calculated. The Tukey test was used to compare alpha diversity metrics among groups. For beta diversity analysis, non-metric multidimensional scaling (NMDS) was performed in R (using vegan, and ggplot2 packages) to visualize differences and similarities of the bacterial composition between the different sampling locations. Analysis of similarities (ANOSIM) was performed in R package to quantitatively compare the microbial community differences between different groups. Phylogenetic investigation of communities by reconstruction of unobserved states(PICRUSt) (Langille et al. 2013) was then applied to predict the functional profiles of the gut microbial communities, and annotated by using the Kyoto Encyclopedia of Genes and Genomes (KEGG) database.
Metabolomic analyses Metabolites extraction
To investigate the common metabolites in the fecal metabolome of Himalayan Griffons, a total of 12 fecal samples from 4 groups were shipped to the Allwegene Technology Co. Ltd. (Beijing, China) for metabolomic analyses. The sample and group number were reduced because of the cost associated with untargeted metabolomics. Seven milligram of sample was extracted by 500 μL extraction solvent containing an internal target(2:2:1, methanol:acetonitrile:water (v/v/v), which was kept at − 20 °C before extraction). After homogenization for 3 times, incubation for 1 h at − 20 °C to precipitate proteins. The extracts were then dried in a vacuum concentrator without heating, and added 100 μL extraction solvent (v
:v
= 1:1) to reconstitute. The supernatants were carefully collected and transferred into a fresh 2 mL Liquid Chromatography–tandem Mass Spectrometry (LC–MS) glass vial for the ultra-high performance liquid chromatography coupled with a hybrid quadrupole time-of-flight mass spectrometry (UHPLCQTOF-MS) analysis.LC–MS acquisition, data preprocessing, and annotation
LC–MS analyses were performed using an UHPLC system (1290, Agilent Technologies, Santa Clara, CA, USA)with a UPLC BEH Amide column (1.7 μm, 2.1 × 100 mm,Waters) coupled to TripleTOF 6600 (AB SCIEX, Foster City, CA, USA). The mobile phase consisted of 25 mM NHOAc and 25 mM NHOH in water (pH = 9.75; A) and acetonitrile (B) was carried with elution gradient as follows: 0 min, 95% B; 7 min, 65% B; 9 min, 40% B; 9.1 min,95% B; 12 min, 95% B, which was delivered at 0.5 mL per minute. The injection volume was pos: 1 μL, and neg:2 μL. The Triple TOF mass spectrometer was used for its ability to acquire MS/MS spectra on an informationdependent basis (IDA) during an LC/MS experiment. In this mode, the acquisition software (Analyst TF 1.7, AB SCIEX, Foster City, CA, USA) continuously evaluates the full scan survey MS data as it collects and triggers the acquisition of MS/MS spectra depending on preselected criteria. In each cycle, 12 precursor ions whose intensity greater than 100 were chosen for fragmentation at collision energy (CE) of 30 V (15 MS/MS events with product ion accumulation time of 50 ms each). The electrospray ionization (ESI) source conditions were set as following:ion source gas 1 as 60 Psi, ion source gas 2 as 60 Psi, curtain gas as 35 Psi, source temperature 650 °C, Ion Spray Voltage Floating (ISVF) 5000 V and − 4000 V in positive and negative modes, respectively. MS raw data (.wiff) files were converted to the mzXML format using ProteoWizard, and processed by R package XCMS (version 3.2). The preprocessing results generated a data matrix that consisted of the retention time (RT), massto-charge ratio(m/z) values, and peak intensity. R package CAMERA was used for peak annotation after XCMS data processing. In-house MS2 database was applied in metabolites identification.
Isolation, and identification of gut bacteria Culture media
Four types of culture media were used in this study for isolation of gut bacteria, including Luria–Bertani (LB)medium, deMan-Rogosa-Sharpe (MRS) medium, M17 medium, andLactobacillus
selection (LBS) medium.These media are commercially available in Hope Bio-Technology Co. Ltd. (Qingdao, China). Anaerobic culturing was incubated at 37 °C in a A35 anaerobic workstation (Don Whitley, Yorkshire, UK).Sample treatment and bacterial isolation
The fecal samples of Himalayan Griffons were blended and shaken well in phosphate-buffered saline. The suspension was further diluted into different concentrations,and 100 μL of each dilution was plated onto agar plates for incubation at 37 °C under aerobic or anaerobic conditions.The single colonies appearing on the agar plates after incubation for 16 h and 40 h were picked. Single-colony isolation was repeated at least five times for purification of each of the bacteria isolates. Finally, all isolated strains were stored in 30% (v/v) glycerol (Bacterial fluid:100%glycerol = 7:3, v/v) at − 80 °C for further study.
Identification of bacterial isolation
All the isolates were confirmed and identified by sequencing the 16s rRNA gene. Microbial DNA was extracted using the QIAamp DNA Stool Mini Kit (Qiagen, Hilden, Germany) following the manufacturer’s protocol. The universal PCR primers 27F (5′-AGT TTG ATCMTGG CTC AG-3′) and 1492R (5′-GGT TAC CTT GTT ACG ACT T-3′) were used to amplify the 16S rRNA gene. The PCR reaction were performed in 25 μL mixture containing 2.5 μL of 10× Buffer (with Mg), 0.5 μL of each universal primers, 1 μL of dNTP (2.5 mM), 15 μL of sterilized water, and 0.5 μL of genomic DNA. The PCR products were checked for the expected size on 1.0%agarose gel, and then identified using Sanger sequencing by Sangon Biotech Company in Shanghai, China. All the 16S rRNA gene sequences were subjected to BLAST analysis on the NCBI website. The phylogenic tree was constructed using the neighbor-joining (NJ) method with MEGA X software (Kumar et al. 2018).
Antibacterial activity by Oxford cup method
Five pathogenic strains, namely,Escherichia coli
ATCC8099,Salmonella enteritidis
CMCC 50041,Staphylococcus aureus
ATCC6538,Shigella dysenteriae
CMCC(B)51105, andPseudomonas aeruginosa
CMCC(B)10104 were purchased from Yitian Biotechnology Co. Ltd. (Beijing, China). These pathogenic strains were aerobically incubated in LB medium at 37 °C and were utilized as indicator bacteria for the antimicrobial tests. The ability of the seven isolated potential gut beneficial strains to resist these pathogenic bacteria was determined by the Oxford cup method (Zhai et al. 2015),with some adjustments. First, each strain was diluted to achieve a concentration about 1 × 10CFU/mL. Then,each pathogenic strain was swabbed evenly on the LB agar plates using sterilized cotton swabs. The three sterilized Oxford cups were situated on the plates and 200 μL of each potential gut beneficial bacteria solution was added in each cup and were incubated at 37 °C for 24 h.After incubation, the transparent area across the cup was considered as the inhibition zone, and the diameter (mm)was measured using a vernier caliper thrice and stated as average ± standard deviation.Data accessibility
Raw sequence reads of 28 fecal samples and 8 carrion’s swabs are publicly available from the NCBI Sequence Read Archive under the accession number PRJNA723140.A total of 13 isolated strains’ 16S rDNA sequences have been deposited in the GenBank database under accession numbers: MT740347, MT740350, MT740351,MT740353, MT740355, MT740378, MT740354,MW092231, MW092229, MW092226, MW092514,MW092228, MW092227.
Results
Diversity indices
The estimated alpha diversity indices (Chao1 and Shannon) did not show differences among the eight groups(Fig. 2). Beta diversity was calculated to evaluate the degree of dissimilarity in bacterial composition between the different sampling locations, using the Bray–Curtis based non-metric multidimensional scaling (NMDS)analysis. Results showed that samples from different locations were broadly indistinguishable and clustered together (Fig. 3). In addition, this clustering pattern was confirmed by an analysis of similarities (ANOSIM)(Additional file 1: Table S2). A high level of similarities in microbial composition were observed in the pairwise comparison groups.
Bacterial community composition in Himalayan Griffons’rotten foods
On the Qinghai-Tibetan Plateau, dead domesticated yaks, Tibetan sheep, Tibetan dogs, and horses make up the main diets of Himalayan Griffons (Additional file 2:Fig. S1). In addition to these domestic animals, some wildlife carcasses were also eaten by Himalayan Griffons,such as the Himalayan Marmot (Marmota himalayana
)we encountered in our field sampling and bird surveys(Additional file 2: Fig. S1). In order to detect the bacterial diversity in the scavenging diets of Himalayan Griffons,sterile swabs were collected from the surfaces of eight different animal carcasses being eaten by the Himalayan Griffons at eight sampling sites. Then, the bacterial diversity of these rotten foods was analyzed by extracting the total genomic DNA from each sterile swab, and sequencing the 16S rRNA gene V4-V5 hypervariable regions using the Illumina MiSeq platform. The results showed that these scavenging diets were mainly dominated by Bacteroidetes, Firmicutes, Proteobacteria, Fusobacteria,and Actinobacteria at the phylum level (Fig. 4a). The proportions of these five phyla in the total bacteria in each rotten food ranged from 77.25 to 99.50%. At the genus level, a total of 19 high abundance genera were identified(Fig. 4b). Among these genera, in addition to the potential bacterial pathogens, three potential probiotic genera (Anaerostipes
,Christensenellaceae_R
-7_group
, andLactobacillus
) were also detected, indicating that not all the bacteria in the rotten foods were pathogenic bacteria. The proportions of these top 19 genera varied largely across the samples reflecting the diversity of bacteria in different rotting carcasses. In addition, the results contained unidentified genera ranging from 0.97 to 30.56%,suggesting that the bacteria in the rotten foods had yet to be explored. Furthermore, a total of 18 OTUs were found to be present in all the rotten foods, and were considered as the core microbiota of Himalayan Griffons’ rotten foods in this study (Additional file 2: Fig. S2).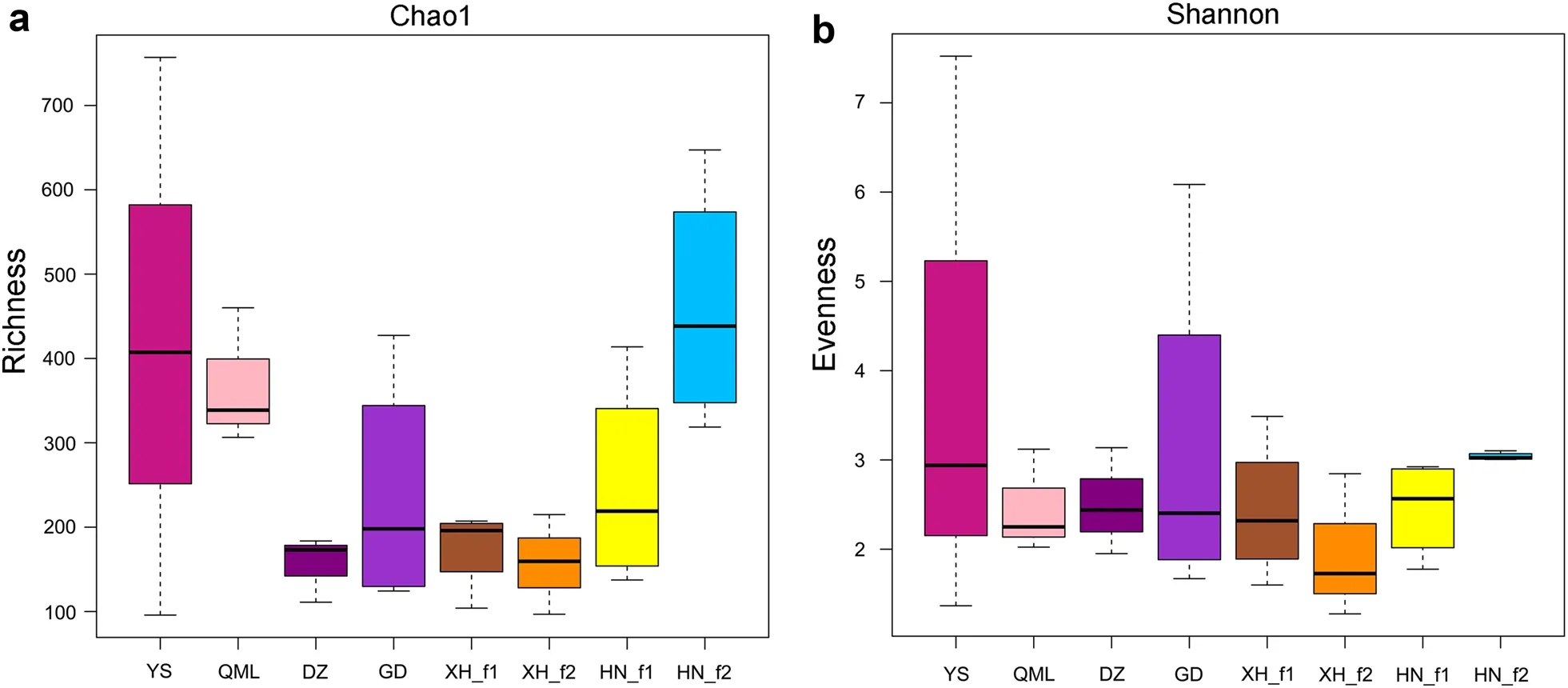
Fig. 2 Boxplots representing alpha diversity metrics of (a) richness (Chao1 index) and (b) evenness (Shannon index) in the gut microbiota of Himalayan Griffons from eight groups
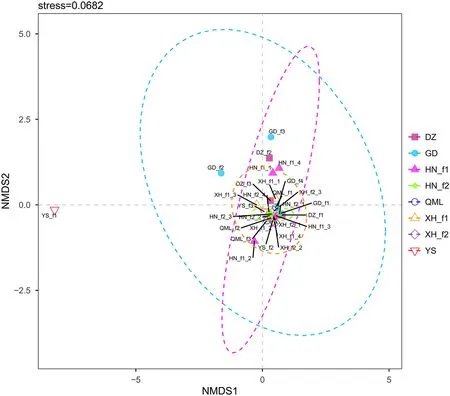
Fig. 3 Non-metric multidimensional scaling (NMDS) of the dissimilarity in the 28 fecal samples from 8 groups
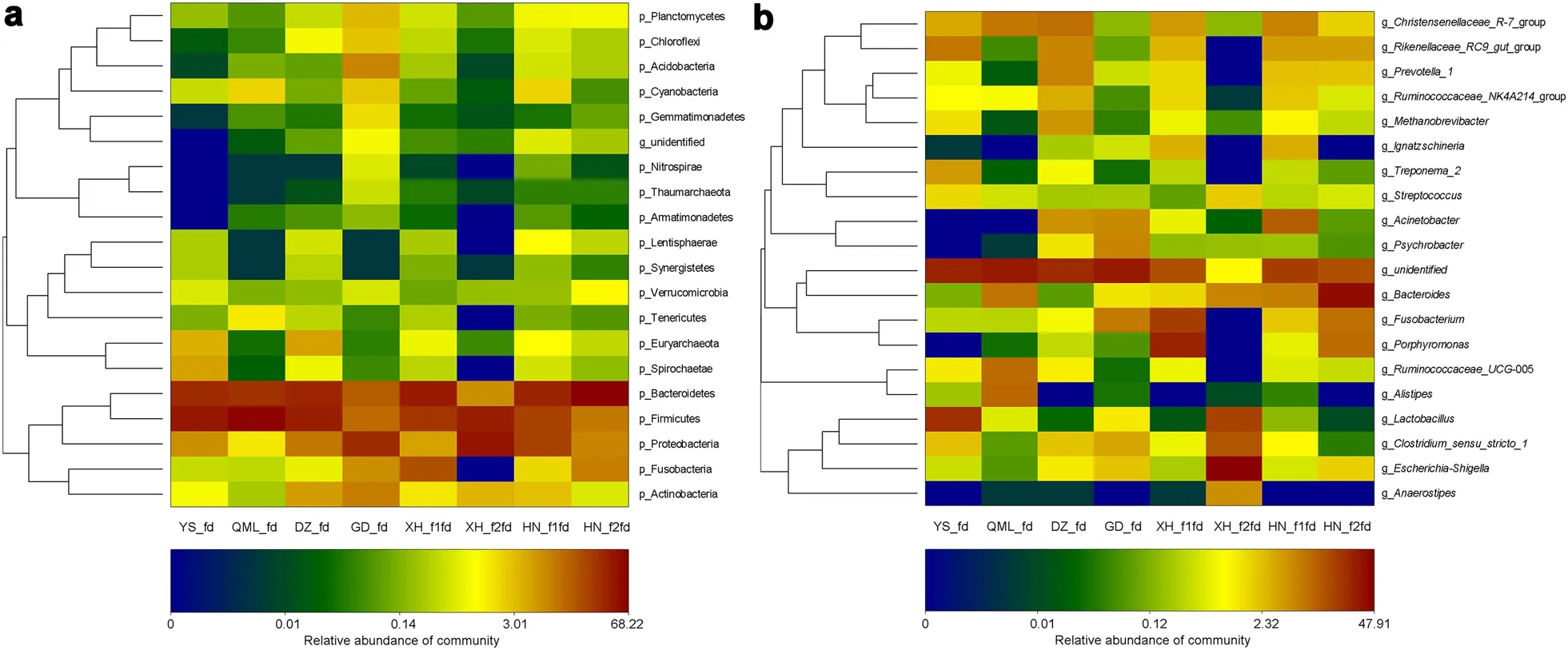
Fig. 4 Heat map of top 20 phyla (a) and genera (b) of each food sample
Bacterial community composition of Himalayan Griffons
A total of 28 fresh fecal samples were collected from Himalayan Griffons foraging flocks at eight different locations on the Qinghai Plateau. We then used amplicon sequencing of the V4–V5 regions of 16S rRNA gene of each sample to identify and characterize the gut microbial community composition of Himalayan Griffons. In total, 1,877,243 raw reads were obtained from all samples (Additional file 1: Table S3). After filtration, 1,801,205 high-quality sequences were produced,with the number of sequences per sample ranged from 30,336 to 175,653 (Additional file 1: Table S3). These high-quality reads were then subsampled to an equal sequencing depth (28,624 reads per sample), and were clustered into a total of 1638 OTUs using a sequence similarity of 97% (Additional file 1: Table S3). The Good’s coverage ranged between 99.48 and 99.93%,indicating that the majority of microbial phylotypes in each sample were detected in this study (Additional file 1: Table S3). The species accumulation curve was also performed to determine whether our 28 fecal samples provided enough OTU coverage for accurate description of the microbial composition present in Himalayan Griffons. As shown in Additional file 2: Fig.S3, the number of OTUs increased quickly at the range of 1‒19 samples, and reached a plateau at the end of our sampling, suggesting that we have largely explored the bacterial composition in this study.
OTUs with the relative abundance greater than 1%were used and classified into different taxonomically levels by the RDP classifier at a 97% confidence threshold.We identified 28 phyla, 76 classes, 103 orders, 192 families, and 393 genera from these OTUs (Additional file 1:Table S4). The composition of microbial community at the levels of Class, Order, and Family is shown in Additional file 2: Figs. S4‒S6.
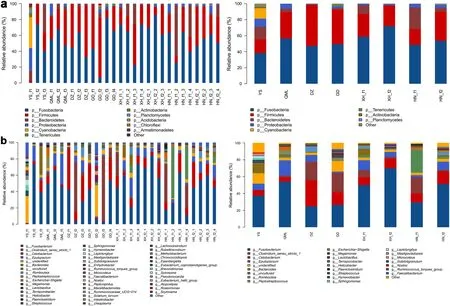
Fig. 5 Relative abundance of the dominant bacterial phyla (a) and genera (b) in each fecal sample and each group, respectively
Relative abundances of the phyla and genera in each sample are summarized in Fig. 5. At the phylum level,the top four abundant phyla in the Himalayan Griffons were Fusobacteria (52.95%), Firmicutes (31.86%),Bacteroidetes (8.62%), and Proteobacteria (3.69%)(Fig. 5a). The proportions of these four phyla comprised more than 97% of the total composition of the samples.At the genus level, the most abundant microbial community wasFusobacterium
(42.45%), followed byClostridium_sensu_stricto_1
(12.96%),Cetobacterium
(10.20%),Epulopiscium
(7.84%), andBacteroides
(4.87%) (Fig. 5b).Another four genera were detected at low levels (relative abundance was between 1 and 2%), includingRomboutsia
(1.88%),Peptostreptococcus
(1.16%),Escherichia
-Shigella
(1.11%), andMegamonas
(1.00%) (Fig. 5b). The proportions of these nine genera comprised more than 83% of the total composition of the samples. In addition, 8.33%of the total sequences were classified to the uncultured and unidentified genera, indicating that the Himalayan Griffons’ gut microbiome might harbor new and not well described microbial species.The “core” gut microbiome of Himalayan Griffons
The core gut microbiomes were investigated at the OTU level across samples. Figure 6 showed the different core thresholds (fraction of 28 samples) and the corresponding number of core OTUs detected. For example, when the core thresholds were set at 100% (present in all the 28 samples), a total of 9 OTUs were identified (Fig. 6; Additional file 1: Table S5). These OTUs accounted for 64.43%of the total reads. The top 3 core OTUs were belonged to the following 3 genera:Fusobacterium
,Clostridium_sensu_stricto_1
, andEpulopiscium
. Using the even less strict core criterion (present in 14 samples, 50% core thresholds), 69 OTUs were totally detected (Fig. 6; Additional file 1: Table S6), and accounted for 90.87% of the total reads. In addition to the above 3 abundant genera,Cetobacterium
was found to be another genus with high abundance (9.64%).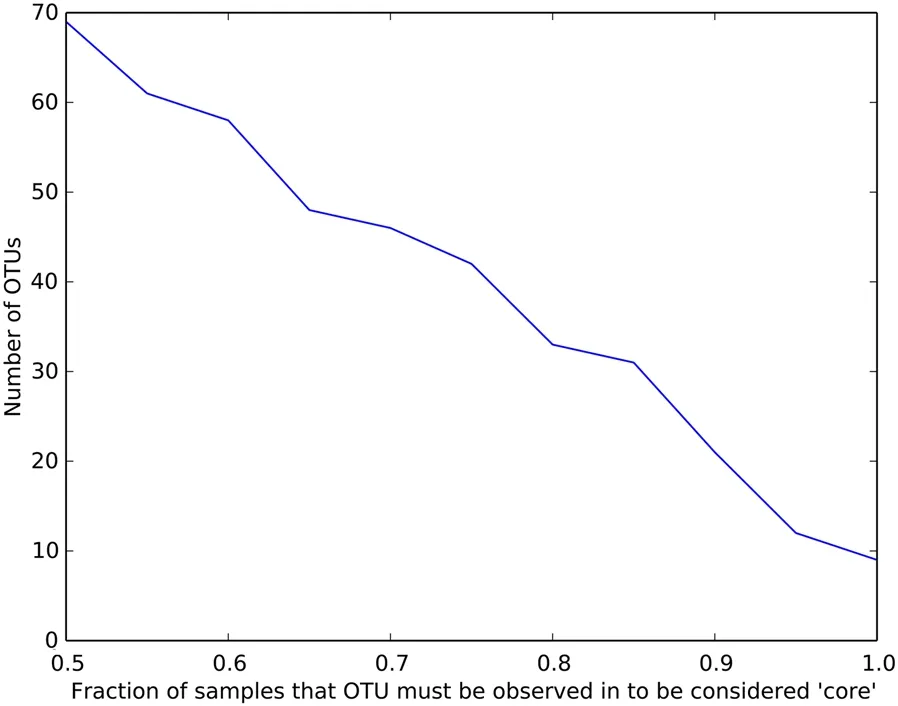
Fig. 6 The number of core OTUs determined by the percentage of occurrence in the sample
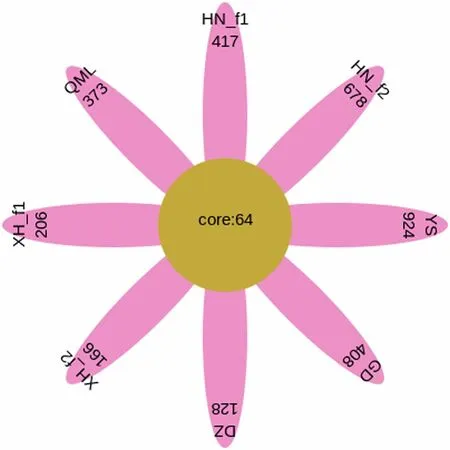
Fig. 7 Flower plot displays the number of common/unique OTUs across different groups
We next examined the core gut microbiomes across different groups. The core 64 bacterial OTUs present in all the groups were identified (Fig. 7; Additional file 1:Table S7). In this case, the core genera with high abundance is still those 4 genera, i.e.,Fusobacterium
,Clostridium_sensu_stricto_1
,Cetobacterium
, andEpulopiscium
.Predictive functional profiling of the Himalayan Griffons’gut microbiome
In addition to providing information on the gut microbial community structure of Himalayan Griffons, we predicted the functional profiles of the Himalayan Griffons’gut microbiome. A total of 302 KEGG pathways at level 3 were predicted (Additional file 3: Table S8). Among these,the largest group was related to metabolism (47.30%),consisting of top 5 higher proportions of carbohydrate metabolism (22.97%), amino acid metabolism (18.65%),energy metabolism (11.66%), metabolism of cofactors and vitamins (9.52%), and nucleotide metabolism (8.78%)(Fig. 8). Collectively, these top 5 functional categories within the metabolism group accounted for 71.58% of the reads. Genetic information processing (18.93%) was the second most abundant functional group, replication and repair (45.04%) was the most abundant functional category within this group, followed by translation (29.29%),transcription (13.65%), and folding, sorting and degradation (12.01%) (Fig. 8). Approximately one third of the sequences in the replication and repair category were classified into DNA repair and recombination proteins(KEGG level 3) (32.70%). The third most prominent functional group was environmental information processing(15.33%), including the categories of membrane transport(87.98%), signal transduction (10.36%), and signaling molecules and interaction (1.66%) (Fig. 8). The vast majority of reads within the membrane transport category were distributed in transporters (51.30%) and ABC transporters (27.35%). Furthermore, a total of 263 KEGG pathways (level 3) were found to be present in all the 8 groups, which could be considered as core functions of Himalayan Griffons’ gut microbiome (Additional file 2:Fig. S7; Additional file 4: Table S9).
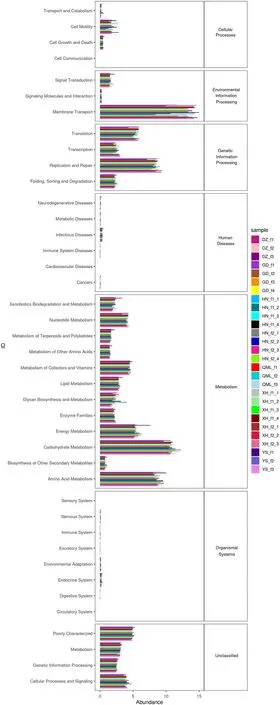
Fig. 8 The predicted KEGG metabolic pathways for each sample
Metabolite profiles of the Himalayan Griffons’ gut microbiome
LC–MS metabolomic analysis of the 12 fecal extracts of Himalayan Griffons detected a total of ~ 4490 metabolites (Additional file 5: Table S10, Additional file 6:Table S11). This was relatively comprehensive information of metabolite profiles for this species, which,to the best of our knowledge, has not been reported previously. As shown in Additional file 7: Table S12, a total of 154 metabolites were identified in all the samples. Among these metabolites, the top 15 metabolites accounted for 52.9% of the total metabolites, including some metabolic compounds that have important physiological effects, such as 2-methylbutyroylcarnitine,3-(phosphoacetylamido)-L-alanine, adenine, cucurbitacin B, 1-palmitoyl-2-(5-keto-6-octendioyl)-snglycero-3-phosphatidylcholine, 3,5-dinitro-L-tyrosine,stearoylcarnitine, cholic acid, andN
-acetyl-L-aspartic acid.Isolation and identification of gut bacteria from Himalayan Griffons
To increase the depth of this study, using culture dependent methods, the feces of Himalayan Griffons were subjected to bacterial isolation and identification. Fecal samples were inoculated into four different media and incubated aerobically and anaerobically at 37 °C. The uncultivable bacteria were not considered in this study,we focused only on isolating the easily culturable bacteria from the gut of Himalayan Griffons. A total of 184 bacterial isolates were finally isolated, including 63 isolates from Luria–Bertani (LB) media, 57 isolates from deMan-Rogosa-Sharpe (MRS) media, 33 isolates from M17 media, 31 isolates fromLactobacillus
selection(LBS) media. All the isolated bacteria were identified by full-length 16S rRNA gene sequence analysis, and then compared by the NCBI GenBank database. These bacteria were classified into 3 different phyla, 8 genera and 13 species. The 13 species’ representative sequences were submitted to NCBI GenBank and got accession numbers (Table 1). The phylogenetic tree of these species was shown in the Additional file 2: Fig. S8.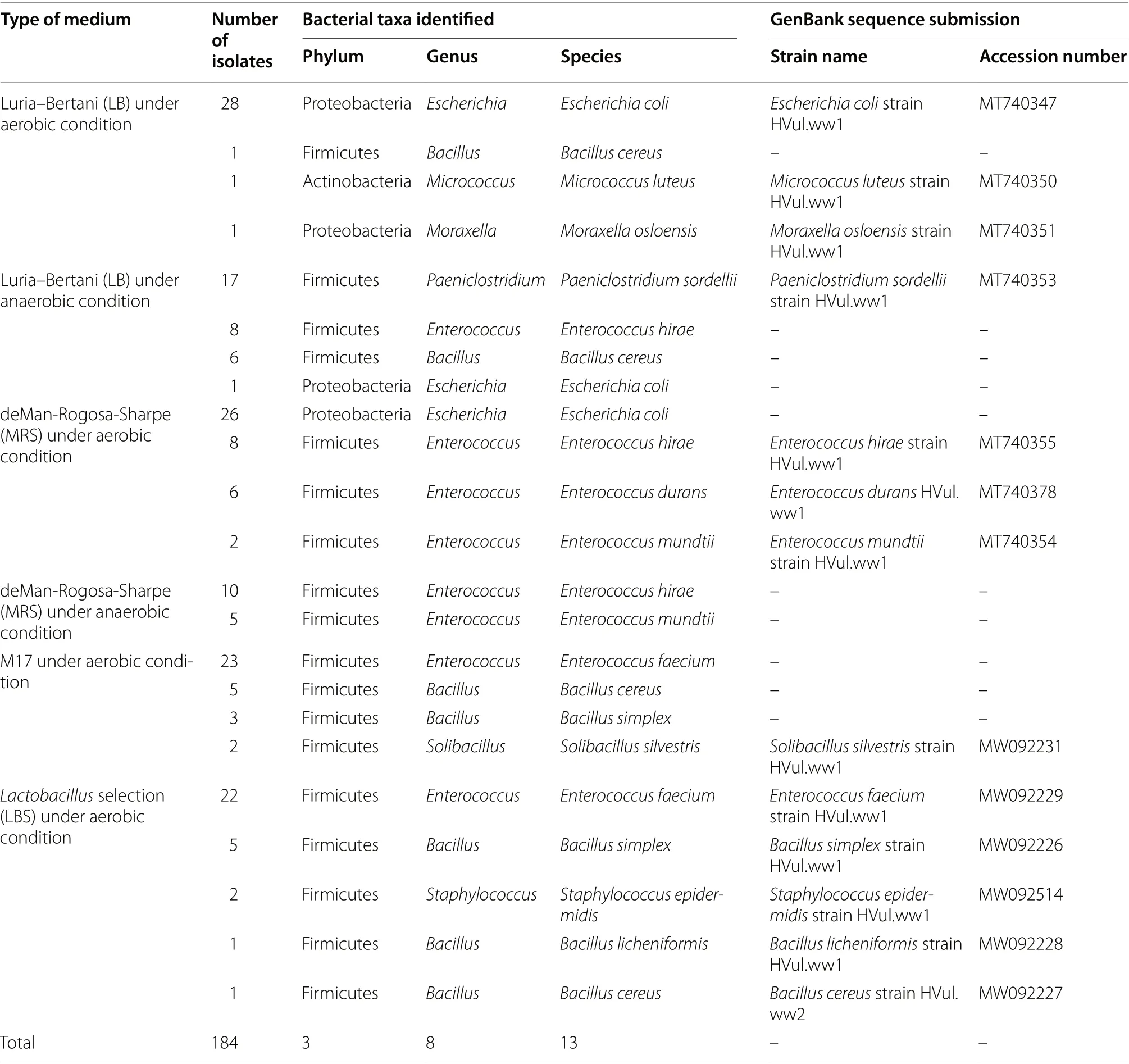
Table 1 Bacteria isolated from various media conditions, and identified based on 16S rRNA gene sequencing analysis
Antagonistic activities of potential gut beneficial bacteria against pathogens
To test whether the potential beneficial bacteria in the gut of Himalayan Griffons assist the host in inhibiting the pathogenic bacteria, the ability of the seven isolated strains (Enterococcus durans
HVul.ww1,Enterococcus hirae
strain HVul.ww1,Enterococcus mundtii
strain HVul.ww1,Enterococcus faecium
strain HVul.ww1,Bacillus cereus
strain HVul.ww2,Bacillus licheniformis
strain HVul.ww1, andBacillus simplex
strain HVul.ww1) to resist five pathogen strains (Escherichia coli
ATCC8099,Salmonella enteritidis
CMCC 50041,Staphylococcus
aureus
ATCC6538,Shigella dysenteriae
CMCC(B)51105,andPseudomonas aeruginosa
CMCC(B)10104) was determined in this study (Fig. 9). All the seven isolated strains showed inhibition activity againstSalmonella enteritidis
andStaphylococcus aureus
in the antibacterial experiment. The inhibition zone diameter ranged from 8.74 to 16.85 mm.Enterococcus durans
HVul.ww1 showed the strongest inhibition ofEscherichia coli
,Salmonella enteritidis
, andPseudomonas aeruginosa
, with a diameter of 16.44 mm, 16.85 mm, and 12.07 mm,respectively (Fig. 9).Enterococcus hirae
strain HVul.ww1 exhibited the strongest inhibitory effect againstShigella
dysenteriae
, with a diameter of 13.91 mm (Fig. 9).Bacillus simplex
strain HVul.ww1 had the strongest inhibitory effect onStaphylococcus aureus
, with a zone of inhibition of 13.29 mm (Fig. 9).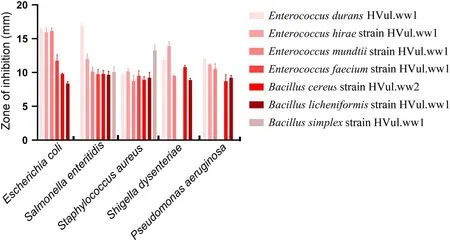
Fig. 9 Antimicrobial activity of seven isolated strains against five pathogen strains
Discussion
Himalayan Griffons are widely distributed avian scavengers in the Qinghai-Tibetan Plateau, which play an important role in maintaining the balance of the Plateau ecosystem. The nature of scavenging makes Himalayan Griffons more vulnerable to pathogens from rotten food.Over the past decade, numerous studies have shown that the gut microbiome, coevolution with the host, played an important role in host metabolism, health, and adaptation to a specific diet and an extreme environment (Wei et al. 2019; Yao et al. 2019; Huang et al. 2021; Levin et al.2021). Therefore, we propose a “achieve two things at one stroke” hypothesis: (a) the gut microbes of Himalayan Griffons are derived from the saprophagous diets, and(b) at the same time these gut microbes assist the host in digestion, absorption, and metabolism of saprophagous diets, and some gut microbes convey resistance to pathogenic bacteria in saprophagous diets. Analysis of the structure and function of gut microbiome in Himalayan Griffons is a key step to test our hypothesis. While, due to the difficulty of collecting samples from wild Himalayan Griffons, this part of the research is still limited. In this study, culture-independent method, using 16S rDNA high-throughput sequencing on 28 fecal samples from 8 flocks, was performed to investigate the gut microbiota composition and function traits. At the same time, the culture-dependent method, using 4 types of media culturing 184 isolates, was used to study the antibacterial activity of gut beneficial bacteria. Integration of cultureindependent and dependent methods provides a more comprehensive picture of the Himalayan Griffons’ gut microbiome, greatly expanding upon previous microbiome profiles analyzed from only 3 rectal swabs of Himalayan Griffons (Meng et al. 2017c).
Gut microbiota structures and functions of Himalayan Griffons
At the phylum level, the gut microbiota of the Himalayan Griffons in the present study was dominated by Fusobacteria, Firmicutes, Bacteroidetes, and Proteobacteria. These results are similar to the findings of previous studies on the gut microbiome of other vultures’species (Roggenbuck et al. 2014; Zepeda Mendoza et al.2018; Meng et al. 2017c) and other carrion feeders (Ley et al. 2008; Keenan et al. 2013), but in different proportions. Firmicutes, Bacteroidetes and Proteobacteria are the most common bacterial phyla present across different kinds of animals’ gut microbiota, while Fusobacteria has frequently been detected in the gut microbiota of carnivorous and omnivorous birds (Waite and Taylor 2015). Fusobacteria was the phylum with the highest abundances in the Himalayan Griffons’ gut microbiota,members in which are often studied in the context of pathogenicity. Overall, Fusobacteria was reported to link to carcinogenesis in multiple cancer types and associated with a range of important chronic diseases (Kelly et al.2018), owing to their ability to colonize or invade through mucosal surfaces and to activate an immune response(Rubinstein et al. 2013). However, Zepeda Mendoza et al.(2018) found that the gut Fusobacteria could provide health protection for the host (the Black Vultures and the Turkey Vultures) in a beneficial manner by the formation of biofilms and colonization resistance against other external pathogens. We speculate that the same protective effect may be also present in the Himalayan Griffons.Further microbial culture and genome studies are needed to isolate the relevant strains in this phylum to reveal the anti-pathogen mechanisms. In this study, the most abundant genus within Fusobacteria wasFusobacterium
andCetobacterium
. Interestingly, genusCetobacterium
isolated from the intestine of freshwater fish was reported to produce vitamin B-12 for the host (Ramirez et al. 2018).Therefore, the potential role of members in this phylum in decomposing carrion and nutrition requires further analysis, which is crucial to testing our hypothesis.The relative abundance of Firmicutes and Bacteroidetes occupied the second and the third place respectively in the gut microbiota of Himalayan Griffons. All these two phyla were considered as symbiotic microbiota essential for the nutrition digestive activity across many birds and mammals. In general, Firmicutes species can benefit their host in digestion and absorbing nutrients by decomposing a range of substances (carbohydrates, polysaccharides, sugars, and fatty acids) via digestive enzymes(Padayachee et al. 2020), whereas Bacteroidetes species usually help the host to degrade complex biopolymers(David et al. 2014). For example, members of Firmicutes have been found to produce high levels of the shortchain fatty acids (SCFAs) as byproducts of fermentation,which can be absorbed directly by the host gut epithelial cells as an energy source (Berry 2016). In addition,gut Firmicutes was found to be involved in T-lymphocyte immunity in chickens (Simon et al. 2016), which could indicate a possible role of these bacterial phyla in adaptive immune function of Himalayan Griffons. The abundant genera identified within Firmicutes included
Clostridium_sensu_stricto_1
,Epulopiscium
,Romboutsia
,Peptostreptococcus
, andMegamonas
. Among these genera,Clostridium_sensu_stricto_1
andRomboutsia
were found to be positively associated with amino acid metabolism (alanine, phenylalanine, and glutamine) and energy metabolism (citrate, pyruvate, creatine, and lactate) in serum (Gao et al. 2019).Epulopiscium
is a morphologically diverse group of intestinal symbionts, and contributes to breakdown of refractory algal polysaccharides ingested by their hosts (Arroyo et al. 2019). This genus was usually found in high abundance amongst herbivorous surgeonfishes (Miyake et al. 2016), but its presence in the gut of Himalayan Griffons deserves further study.Peptostreptococcus
andMegamonas
seemed to have an association with pathophysiological processes in several immunological diseases (de Paepe et al. 2017;Barnes et al. 2020). How these pathogens coexist with the Himalayan Griffons remains a question worth studying.Bacteroides
, with the highest percentage within the Bacteroidetes phylum, is usually associated with the consumption of animal fat and protein-rich diets (Wu et al.2011). The possible beneficial role of microbes within this genus linked to the carrion-based diet in vulture species is still lacking. Additionally, higher Firmicutes to Bacteroidetes ratio associated with obesity in humans and rodents was reported to increase the calories uptake effi-ciency from food (Grigor’eva 2021). However, there is a lack of data to compare the ratio of Firmicutes to Bacteroidetes in the gut microorganisms of vultures with those of birds that have other feeding habits.Proteobacteria in much less abundant occupied the fourth place in the gut microbiota of Himalayan Griffons.The most abundant taxa in this phylum was the pathogenicEscherichia
-Shigella
, which was also isolated from other birds (Diakou et al. 2016). It seems that Himalayan Griffons, the nature’s most successful avian scavengers,could be an animal reservoir of many zoonosis that originated from the carrion (Meng et al. 2017c). Studying the bacteriome and virome of this bird species in the future will provide important information to predict or prevent infection outbreaks.In this study, PICRUSt was used to analyze the microbial functions of Himalayan Griffons. Results showed that the most abundant functional classes were associated with carbohydrate and amino acid metabolism, replication and repair, and membrane transport. Carbohydrate and amino acid metabolism pathway in the gut microbiota may play an important role in providing nutrients to Himalayan Griffons by digesting, and assimilating the carrion. Replication and repair pathway, especially DNA repair and recombination, was much higher in the gut microbiota of Himalayan Griffons, which may be induced by the increased threats and harmful substances from carrion-born pathogens. The enriched membrane transport pathway likely reflects the uptake of more nutrient substrates for direct microbial utilization, as indicated by the chicken gut metagenome (Huang et al. 2018). These data facilitate understanding of the relationship between gut microbial taxa and metabolism, as well as the influence of gut microbes on host physiology.
Core gut microbiota and metabolites of Himalayan Griffons
In this study, NMDS clustering results exhibited larger overlap, and suggested that the majority of gut microbiota between all the samples were conserved. The specialized and conserved saprophagous habit was considered as potential cause for this clustering pattern (Huang et al.2021). Furthermore, using the strict core (100% of the 28 samples, and 100% of the 8 groups), and the relaxed core (50% of the samples), the top 4 abundant core genera were detected, includingFusobacterium
,Clostridium_sensu_stricto_1
,Cetobacterium
, andEpulopiscium
. These core genera of the Himalayan Griffons’ gut microbiome overlapped with the core genera of the different carrion microbiome, which also supports our hypothesis about the carcass’s origin of the Himalayan Griffons’ gut microbiome. These core gut microbiome might form a symbiotic relationship with Himalayan Griffons after long-term co-evolution, thus playing essential roles in many physiological activities of Himalayan Griffons.The metabolic composition of fecal extracts from Himalayan Griffons provides a chance to elucidate the complex metabolic interplay between the host and their gut microbiome. Fecal metabolome consisted of various metabolites with a wide range of physiological properties.In this study, we observed a total of 154 metabolites present in all the 12 fecal samples. The top 15 metabolites accounted more than half of the total metabolites, and are worthy of further physiological research. For example, 2-methylbutyroylcarnitine, stearoylcarnitine, and cholic acid are associated with the metabolism of fatty acids (Ghonimy et al. 2018). Carnitine can be synthesized in animals from the amino acids’ lysine, but cannot be de novo synthesized by any bacterial species (Meadows and Wargo 2015). Interestingly, this substance is an ideal compatible solute that can be utilized by bacteria to protect themselves against stresses such as salt, pressure, temperature, and so on (Smiddy et al. 2004). The presence of 3-(phosphoacetylamido)-L-alanine, Adenine,and 3,5-dinitro-L-tyrosine may also reflect the involvement of gut microbiota in the catabolism of protein and nucleotide in the intestinal lumen. Cucurbitacin B and its close relatives were reported to have potent anticancer activities (Garg et al. 2018). The presence of this metabolite suggested that the gut microbes and their metabolites in the Himalayan Griffons are a potential source of therapeutically important compounds worthy of further exploration. Due to the small sample size of our metabolome, the results were only a preliminary data of the metabolome of the Himalayan Griffons and their gut microbiome. In the future, a larger sample size (at least 10 samples in each group) will be needed to obtain a broad picture of the Himalayan Griffons’ gut metabolome and to conduct comparative analysis among different groups.
Bacterial isolates and the inhibition activity against pathogens
As a supplement to the culture-independent method, culture-dependent method was also used in this study, and might provide a powerful strategy to study the function of specific strains from the Himalayan Griffons. However,usually less than 1% of the gut microbes can be cultured,and the culture-dependent method often produce biased results. Therefore, the culturable gut bacteria in our study were likely to be the high abundant and most active bacterial group in the Himalayan Griffons. In total, 184 culturable gut bacteria were isolated using 4 different types of media and assigned into 3 phyla, 8 genera, 13 species,dominated byEscherichia coli
(29.89%),Enterococcus faecium
(24.46%),Enterococcus hirae
(14.13%), andPaeniclostridium sordellii
(9.24%). Due to the influence of the isolation media on the preference of culturable gut bacteria, more types of media should be used to obtain more culturable bacterial isolates in the future. In addition, culturomics (high-throughput culture) (Lagier et al. 2012)need to be applied to culture the Himalayan Griffons’gut microbiome in order to better identify more culturable microorganisms. Culturomics allows for a wide variety of maintenance and enrichment conditions (culture medium composition, temperature, incubation time,antibiotics, and heat elimination of non-spore forming bacteria, etc.) to cover the majority of microbial diversity in the sample (Pinevich et al. 2018). This study focuses on the isolation and culture of gut beneficial bacteria from Himalayan Griffons, with the aim of testing our hypothesis that gut beneficial bacteria can assist the host to resist the pathogenic bacteria in their diets. Seven potential beneficial gut bacteria were found to have different degree of inhibitory effect on 5 common pathogenic bacteria. This suggested the importance of gut beneficial bacteria mediated health protection for adaptation of Himalayan Griffons to their scavenging habits. This also indicated that the mechanisms of scavenging adaptation in vulture species may not only lie within their genomes(Chung et al. 2015; Zhou et al. 2019), but also in their associated gut microbes. To date, culturable gut microbiome data for only a few species of vultures have been reported, and tend to be biased toward reporting isolated pathogens (Meng et al. 2017a, b, 2018; Plaza et al. 2019),while reports on probiotics are scarce. Our data also suggested that certain types and numbers of probiotics are present in the Himalayan Griffons’ gut microbiome. In the future, germ-free vultures or other bird species as a model should be experimentally transplanted with these microbes to determine the essential physiological functions of these probiotics. Another interesting finding was that we also found beneficial bacteria in the carrion microbiome. Thus, we speculate that Himalayan Griffons may encounter beneficial bacteria in their diets, and these probiotics were selectively colonized the gastrointestinal tract during evolution.Conclusions
This study is the first to provide comprehensive analyses of the Himalayan Griffons’ gut microbiome using the combination of the culture-independent and culture-dependent methods. Our results showed a high abundance of four major phyla in Himalayan Griffons,including Fusobacteria, Firmicutes, Bacteroidetes, and Proteobacteria. At the genus level, the most abundant microbial community wereFusobacterium
, followed byClostridium_sensu_stricto_1
,Cetobacterium
,Epulopiscium
, andBacteroides
. Further, we found the core gut microbial composition at both sample and group levels.The predicted primary functional categories of the Himalayan Griffons’ gut microbiome were associated with carbohydrate and amino acid metabolism, replication and repair, and membrane transport. Taken together,these results revealed the composition and functions of the gut microbiota in Himalayan Griffons. The first highthroughput results characterizing the microbiota of the different types of carrions could be used as a base for understanding the origins of the Himalayan Griffons’ gut microbes. We also present here the results of the isolation and culture of gut beneficial bacteria, the inhibitory effect on pathogenic bacteria, as well as the fecal metabolome, that suggest gut microbes play a crucial role in the scavenging adaptation of Himalayan Griffons. However,the linkage between the gut microbiota and the carrions utilization and metabolism was not studied in our current work, which may be the further research plan in our laboratory. Thus, based on our results, we envision that future multi-omic studies (including genome, metagenome, virome, and culturome) of Himalayan Griffons should be conducted to more comprehensively explore the mechanisms of scavenging adaptation in this species and to better protect this species.Supplementary Information
The online version contains supplementary material available at https:// doi.org/ 10. 1186/ s40657- 021- 00287-0.
Additional file 1: Table S1. The administrative region, coordinates,altitude, number of samples, and type of animal carrion of each sampling site. Table S2. ANOSIM R-statistics andP
-values between groups.Table S3. Summary of sequencing data of each fecal sample. Table S4.The number of detected bacterial taxonomic units (from phylum level to genus level) in each fecal sample. Table S5. Taxonomic classification and relative abundance of the nine core OTUs when the threshold is set at 100% (n
= 28). Table S6. Taxonomic classification and relative abundance of the 69 core OTUs when the threshold is set at 50% (n
= 28). Table S7.Taxonomic classification and relative abundance of the 69 core OTUs present in all the 8 groups.Additional file 2: Figure S1. Different types of Himalayan Griffons’ food commonly encountered in the fieldwork. Figure S2. Flower plot displays the number of common/unique OTUs across different Himalayan Griffons’foods. Figure S3. Species accumulation curve shows the total number of OTUs in a given number of fecal samples. Figure S4. Relative abundance of gut bacterial taxa at the class level. Figure S5. Relative abundance of gut bacterial taxa at the order level. Figure S6. Relative abundance of gut bacterial taxa at the family level. Figure S7. Flower plot displays the number of common/unique KEGG metabolic pathways across different groups. Figure S8. The phylogenetic tree of the 13 species identified in this study based on the 16S rRNA gene sequences using the neighborjoining method. The phylogenetic tree was statistically evaluated using 1000 bootstrap replicates.
Additional file 3: Table S8. Information on 302 predicted KEGG pathways for each sample.
Additional file 4: Table S9. Information on 263 predicted KEGG pathways(level 3) present in all the eight groups.
Additional file 5: Table S10. Metabolites detected in the fecal metabolomes of QML group (n
= 3) and YS group (n
= 3).Additional file 6: Table S11. Metabolites detected in the fecal metabolomes of HN_f2 group (n
= 3) and XH_f1 group (n
= 3).Additional file 7: Table S12. Information on 154 metabolites identified in all the samples (n
= 12).Authors’ contributions
WW and KS conceived and designed the study. XLG, SSZ, ZMLC and YL conducted the field work. LLZ, JPH, JYH, XL and HXC carried out the analyses. WW prepared the draft of the manuscript, AD and KS revised it. All authors read and approved the final manuscript.
Funding
This research was funded by the National Natural Science Foundation of China(Grant No. 31960277); the National Natural Science Foundation of China and Russian Foundation for Basic Research Cooperative Exchange Project (Grant Nos. 32111530018, 21-54-53031); the Natural Science Foundation of Qinghai Province of China (Grant No. 2018-ZJ-932Q). Dr. Wen Wang was supported by“1000 Talent” programs of Qinghai Province.
Availability of data and materials
Raw sequence reads of 28 fecal samples and 8 carrion’s swabs are publicly available from the NCBI Sequence Read Archive under the accession number PRJNA723140. A total of 13 isolated strains’ 16S rDNA sequences have been deposited in the GenBank database under accession numbers: MT740347,MT740350, MT740351, MT740353, MT740355, MT740378, MT740354,MW092231, MW092229, MW092226, MW092514, MW092228, MW092227.Other data used in the present study are available from the corresponding author on reasonable request.
Declarations
Ethics approval and consent to participate
This study conformed to the guidelines for the care and use of experimental animals established by the Ministry of Science and Technology of the People’s Republic of China (Approval number: 2006-398). The research protocol was reviewed and approved by the Ethical Committee of Qinghai University. This study did not involve capture or any direct manipulation or disturbance of wild Himalayan Griffons in the fieldwork.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Author details
State Key Laboratory of Plateau Ecology and Agriculture, Qinghai University,Xi’ning 810016, China.College of Agriculture and Animal Husbandry, Qinghai University, Xi’ning 810016, China.College of Finance and Economics, Qinghai University, Xi’ning 810016, China.Xining Wild Animal Epidemic Disease Monitoring Station, Xi’ning 810001, China.College of Eco-Environmental Engineering, Qinghai University, Xi’ning 810016, China.Institute of Systematics and Ecology of Animals, Siberian Branch of the Russian Academy of Sciences, Novosibirsk 630091, Russia.Federal Research Center of Fundamental and Translational Medicine, Novosibirsk 630117, Russia.
Received: 29 April 2021 Accepted: 30 September 2021
杂志排行

Avian Research的其它文章
- Taxonomic revision of the Savanna Nightjar (Caprimulgus affinis) complex based on vocalizations reveals three species
- Taxonomic status of grey-headed Yellow Wagtails breeding in western China
- The composition of mixed-species flocks of birds in and around Chitwan National Park,Nepal
- True grit: ingestion of small stone particles by hummingbirds in West Mexico
- Stopover behavior of Red-eyed Vireos (Vireo olivaceus) during fall migration on the coast of the Yucatan Peninsula
- Phylogeography and morphometric variation in the Cinnamon Hummingbird complex: Amazilia rutila (Aves: Trochilidae)