Phylogeography and morphometric variation in the Cinnamon Hummingbird complex: Amazilia rutila (Aves: Trochilidae)
2022-01-07MelisazquezpezNandadevirtesRodrguezSahidRoblesBelloAlfredoBuenoHernndezLuzZamudioBeltrKristenRueggandBlancaHernndezBaos
Melisa Vázquez-López, Nandadevi Córtes-Rodríguez, Sahid M. Robles-Bello, Alfredo Bueno-Hernández,Luz E. Zamudio-Beltrán, Kristen Ruegg and Blanca E. Hernández-Baños*
Abstract
Keywords: Amazilia rutila, Genetic structure, Genetic variation, Phylogeography, Tres Marías Islands, Trochilidae,Tropical deciduous forest
Background
The Mesoamerican dominion is the area where the Neartic and Neotropics overlap, and includes most of the Mexican and Central American lowlands as well as the Mexican Transition Zone (Morrone 2014). This biogeographic dominion and surrounding areas are well known for possessing high levels of species richness, which are thought to have originated from its complex topography,wide range of environmental conditions, and climatic history (Savage 1966). One of the most distinctive habitats found in the Mesoamerican dominion is the tropical deciduous forest, which has a high number of endemic species (Gordon and Ornelas 2000).
Tropical deciduous forest has the driest conditions of the tropical forests, and precipitation is strongly seasonal (Rzedowski 1978; Meave et al. 2012). In addition, it is considered one of the most threatened habitats in the Neotropics due to land use change, habitat fragmentation, and human population growth (Lerdau et al. 1991;Koleff et al. 2012). The diversity of the tropical deciduous forest has been attributed to historical factors such as the climatic fluctuations of the Pleistocene, in which colder and drier periods promoted its fragmentation, followed by periods of expansion with warmer and more humid conditions (Werneck et al. 2011).
Some studies of bird species distributed in Mesoamerican deciduous forests have shown high levels of genetic and morphological variation associated with historic and demographic events (e.g. Arbeláez-Cortés et al.2014), while others have focused on allopatric populations, where long-term isolation, moderate or low gene flow, presence of geographic barriers, and historical events explained the overall geographic structure (e.g.Smith et al. 2011). One clear example of allopatric differentiation is the taxa inhabiting the Tres Marías Islands(Nayarit, Mexico), where there are several endemic subspecies (Cortés-Rodríguez et al. 2008; Smith et al.2011; Montaño-Rendón et al. 2015). These subspecies share the common feature of clear morphological differentiation, mainly in body size, from their continental counterparts, representing independent evolutionary lineages with defined genetic structure (Ortíz-Ramírez et al. 2018). In addition to the Tres Marías Islands, the Yucatán Peninsula and the Coastal Plain of southwestern Mexico have been considered to have high potential for diversification (García-Deras et al. 2008; González et al.2011; Smith et al. 2011; Ramírez-Barrera et al. 2018;Hernández-Baños et al. 2020). Furthermore, a study based on molecular data showed that species distributed in the Neotropical lowlands have a low and constant rate of diversification over time; extrapolating those rates of diversification to the present leads to a greater number of lineages than is currently described (Weir 2006). The number of species present in the tropics is thus not properly reflected in the currently accepted taxonomy.
Hummingbirds belong to one of the most diverse bird families (Trochilidae: 361 spp.; Gill et al. 2021), with specialized nectarivorous feeding mode and high rates of speciation. This has resulted in a huge diversity of bill morphologies and color patterns among nine highly supported groups (Bleiweiss et al. 1997; McGuire et al. 2014).In this study, we examined the evolutionary history of the Cinnamon Hummingbird (Amazilia rutila
), a mediumsized hummingbird that inhabits tropical deciduous forests ranging from 0 to 1600 m of elevation (Howell and Webb 1995). This species is mainly distributed among three biogeographic provinces: the Pacific lowlands,Yucatán Peninsula, and Mosquito, based on the regionalization of the Neotropical region (Smith 1941; Ryan 1963; West 1964; Morrone 2014). It is a resident species from Sinaloa in northwestern Mexico, south along the Pacific Coast (including the Tres Marías Islands) to Costa Rica, and along the coastal plain of the Yucatán Peninsula south to Belize (including “offshore cays”) and northeastern Nicaragua (Howell and Webb 1995). Based on morphology and allopatric distributions, four subspecies are recognized (Billerman et al. 2020; Gill et al. 2021).A. r. rutila
(DeLattre 1843) is distributed in western and southern Mexico (from Jalisco to Oaxaca) and has green plumage on the upperparts, cinnamon underparts and a rufous tail.A. r. diluta
(van Rossem 1938) inhabits northwestern Mexico (from Sinaloa to Nayarit); its upper parts are more golden-bronze and underparts are paler and less reddish thanrutila
.A. r. corallirostris
(Bourcier and Mulsant 1846) is distributed from south and southeastern Mexico (from Chiapas to the Yucatán Peninsula) to Costa Rica and is much more deeply colored thanrutila
.Finally, located within the Pacific lowlands province, the subspeciesA. r. graysoni
(Lawrence 1866) inhabits the Tres Marías Islands; it is darker in color and larger thanrutila
(Ridgway 1901). In general, the systematics ofA.rutila
are unresolved and controversial. Weller (1999)considered thatA. rutila
forms a superspecies withA.yucatanensis
(Buff-bellied Hummingbird), and in a multilocus molecular study, the genusAmazilia
formed a polyphyletic group nested with the generaHylocharis
,Chrysuronia
,Lepidopyga
, andDamophila
(McGuire et al. 2007).The aims of this study were to: (1) analyze the genetic variation and morphometric differentiation across the geographic distribution ofA. rutila
, (2) reconstruct phylogenetic relationships and past scenarios throughout the geographic range to analyze divergence times and demographic changes over time and space, and (3) propose an evolutionary history and taxonomic hypothesis of intraspecific lineages. We expected to find high levels of genetic structure associated with the isolation of allopatric populations and environmental conditions over continuous ranges.Methods
Sampling, PCR and sequencing
We obtained 84 tissue samples from theA. rutila
complex and one sample fromA. yucatanensis
as the outgroup (see Additional file 1: Tables S1, S2). We supplemented our sampling with GenBank sequences fromA. rutila
,A. yucatanensis
andA. tzacatl
(McGuire et al.2014). Tissues samples were provided by the collection of the Museo de Zoología (MZFC) “Alfonso L. Herrera”(Universidad Nacional Autónoma de México), the Burke Museum (University of Washington), and the Biodiversity Institute (University of Kansas).DNA was extracted using the Qiagen DNAEasy kit,following the manufacturer’s protocols. We amplified and sequenced two mitochondrial markers: partial and complete NADH dehydrogenase subunit 2(ND2: 605–1041 bp), and the full length of cytochrome C oxidase subunit I gen (COI: 568 bp). We also amplified two nuclear markers: the regions between exons 4 and 5 of the Muscle Skeletal Receptor Tyrosine Kinase gene (MUSK: 628 bp), and a segment comprising the end of exon 6 and the beginning of exon 8 of the Ornithine Decarboxylase gene (ODC: 571). We used primers L5219, H6313, L5758 and H5766 for the amplification of ND2 (Sorenson et al. 1999); primers F1-COI and R2-COI for COI (Chaves et al. 2008); ODC2-F and ODC2-R for ODC (McGuire et al. 2007); and MUSK R3 and MUSK F3 for the amplification of MUSK (McGuire et al.2007). All PCR products were confirmed on 1% agarose gel, and sequencing reactions were performed by the Washington University Genomics Center. All new sequences have been deposited in GenBank (Accession numbers MZ998668-MZ998740, OK624605-OK624657,OK614044-OK614080, OK618334-OK618361).
Sequences were proofread using Sequencher v.4.8(Gene Codes Corporation 2007), and aligned with the Clustal W function in BioEdit (Thompson et al. 1994;Hall 1999). We corroborated the mitochondrial origin of our sequences using at least two of the following methods: amplifying/sequencing overlapping gene segments,amplifying/sequencing one region with different primer sets, and sequencing both DNA strands. We found no evidence of contamination of mtDNA sequences.
Phylogenetic analyses, genetic structure and neutrality tests
We used the Bayesian approach implemented in PHASE v.2.1 (Stephens et al. 2001; Stephens and Scheet 2005)to reconstruct haplotypes and avoid heterozygotes in nuclear sequences, selecting the pairs of haplotypes with a posterior probability higher than 0.90. We obtained the models of evolution for each gene using Partition-Finder (Lanfear et al. 2017), using the following parameters: linked branched lengths, greedy search algorithm(Lanfear et al. 2012), and the Bayesian Information Criterion (BIC) for model selection. The phylogenetic analyses were conducted using each locus separately and using the concatenated matrix including nuclear and mitochondrial loci (multilocus). We conducted phylogenetic analysis under Bayesian Inference (BI), using MrBayes 2.0 (Huelsenbeck and Ronquist 2002) in CIPRES Science Gateway (Miller et al. 2010). Each analysis consisted of four independent chains, random starting trees, and uniform prior distribution of parameters. The chains were run for 30 million generations, sampling trees every 1000 generations. The asymptote was determined visually, 5000 burn-in trees were discarded, and the remaining trees from the plateau phase were used to estimate Bayesian posterior probabilities. We considered that clades were strongly supported if they were present in ~ 95% of the sampled trees (Huelsenbeck and Ronquist 2002; Wilcox et al. 2002). Also, a Maximum Likelihood analysis was conducted using RaxML v.8.0.0 (Stamakis 2014) in CIPRES Science Gateway (Miller et al. 2010),using separate partitions for each locus (see “Results”),with 100 bootstrap replicates.
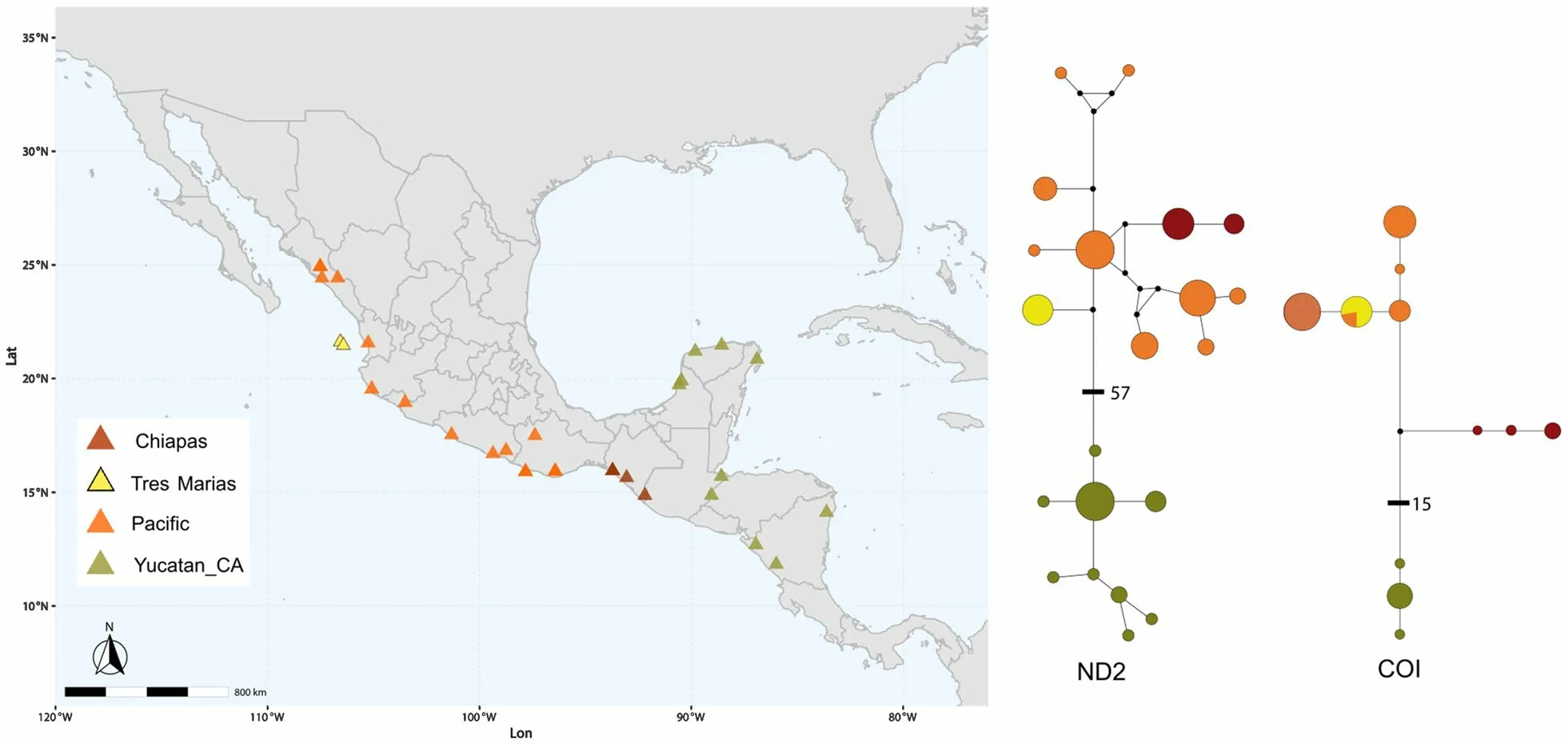
Fig. 1 Geographic location of Amazilia rutila samples used for genetic analyses (left). Haplotype networks of single ND2 and COI mtDNA markers(right). Different colors represent different groups based on haplotype networks and allopatric populations: Pacific: Pacific coast, Tres Marías: Tres Marías Islands, Yucatán_CA: Yucatán Peninsula and Central America
We defined four different groups, corresponding to allopatric populations from sampled localities ofA.rutila
(see Fig. 1): (1) Tres Marías Islands, (2) Pacific coast, (3) Chiapas, and (4) Yucatán Peninsula and Central America (hereafter: Tres Marías, Pacific, Chiapas and Yucatán_CA). For the comparison between populations, we specified geographic groups based on the main clades recovered from phylogenetic analysis(concatenated-multilocus), and tested for substructure in Pacific and Yucatán_CA groups (see “Results”). A median-joining network was constructed to visualize the structure of populations, the number of haplotypes,their frequencies, and the relationships among individuals using the program Network 4.5.1.0 (Bandelt et al.1999). We tested locus neutrality with Fu’sF
s (Fu 1997)and Tajima’sD
(Tajima 1989) metrics in DnaSP v.5(Librado and Rozas 2009), and obtained the summary statistics for each population. We tested the differentiation hypothesis using an analysis of molecular variance AMOVA and theF
fixation index using Arlequin 3.1 (Excoffier and Schneider 2006). These analyses are useful for observing population structure, estimating population differentiation directly from molecular data,and testing the hypothesis of differentiation (Excoffier et al. 1992). Additionally, we used STRU CTU RE 2.3.2(Pritchard et al. 2000) for population assignments under an admixed and LocPrior model (Hubisz et al.2009), with 10,000 iterations of burn-in and 20,000 MCMC (Markov chain Monte Carlo), for 20 replicates of eachK
value (from 1 to 6). To evaluate STRU CTU RE results, we used the Evano method to chooseK
value by∆K
(Evanno et al. 2005), as implemented on the Structure Harvester website (Earl and vonHoldt 2012).Genetic distances were obtained with MEGA under Maximum Likelihood model, with 100 bootstrap replicates (Stecher et al. 2020).Divergence time estimates
We estimated divergence times with the concatenated matrix using Beast v.2.6.2 (Bouckaert et al. 2019). Different partitions by gene were defined based on Pacheco et al. (2011) for ND2 and COI, Lerner et al. 2011 for ODC, and Ellegren (2007) for MUSK. We employed a strict clock and a constant population model as a tree prior. We chose the strict clock model empirically based on data fit, and the constant population model based on our demographic reconstruction (see “Results”). The analysis was run for 200 million generations, sampling every 1000. We used Tracer v.1.7 (Rambaut et al. 2018)to ensure adequate Effective Sample Size (ESS > 200),which was reached for most statistics, with the exception of prior and probability statistics. With TreeAnnotator v.2.6.2 (Rambaut and Drummond 2013) we discarded the first 25% or trees as burn-in and produced a maximum clade credibility tree with 95% highest probability density intervals. The final tree was visualized in FigTree v.1.4.2(Rambaut 2014).
Morphometric analysis
We took linear measurements of five morphological traits from 210 study skins housed at the American Museum of Natural History (AMNH) and at the Museo de Zoología“Alfonso L. Herrera” (MZFC). We measured wing chord(WC) and tail length (TL) using an Avinet calibrated ruler to the nearest 0.5 mm; also, we measured culmen length (CL), beak width (BW), and beak height (BH) to the nearest 0.1 mm with a Mitutoyo digital caliper.
We assigned each specimen to geographic groups defined based on the haplotype network (see “Results”;Fig. 1). We performed Mann–WhitneyU
tests between sexes within each geographic group, with Bonferroni correction to evaluate sexual dimorphism. We also carried out Kruskall-Wallis test with “geographic group” as the grouping variable to assess geographic variation in the measured characters. Finally, we carried out a PCA to visually examine the structure of variation in multivariate space. All statistical analyses were performed in R (R Core Team 2020).Ecological Niche Modelling under past scenarios and demography
To infer suitability of environmental conditions under past scenarios, we performed projections using Ecological Niche Models. Presence records ofA. rutila
were downloaded from GBIF (Global Biodiversity Information Facility) and accessed from R via rgbif (https:// github.com/ ropen sci/ rgbif; taxon key: 2476417; https:// doi. org/10. 15468/ dl. 7k98v7). These records were cleaned over multiple steps using the following R packages: CoordinateCleaner (Zizka et al. 2019), nichetoolbox (Osorio-Olvera et al. 2016), dplyr (Wickham et al. 2020), and raster (Hijmans 2020). We used the sets of bioclimatic layers (current and past) from WorldClim (Hijmans et al.2005; Braconnot et al. 2007; Booth et al. 2014). Our models for the past were projected onto the Last InterGlacial(LIG: 140–120 kya), the Last Glacial Maximum (LGM: 21 kya), and the Mid Holocene (MH: 6 kya) scenarios. We performed these projections using the continental phylogroups (Pacific coast, Chiapas, and Yucatán and Central America); the Tres Marías group was not included in these analyses due to the low number of occurrence records (n
= 3). The selection of bioclimatic variables was based primarily on a principal component analysis, where we considered the most important variables of the first four principal components (from one to three variables),ensuring that no intercorrelated variables were selected(Pearson correlationr
< 0.75, Additional file 2: Fig. S1).To define the area of accessibility for the species, we used the ellipsenm R package (Cobos et al. 2020) to delimit the area that contained occurrence points across the distribution of the species with a polygon representing a buffer area of 75 km (“M” area). Models were performed in Maxent v.3.4.1 (Phillips et al. 2021) with regularization multiplier = 2 and with 10 replicates of cross-validation with no extrapolation. To binarize outputs, we took into account the logistic threshold from maximum training sensitivity plus specificity.We obtained Bayesian Skyline Plots (BSP) using Beast v.2.6.3 (Bouckaert et al. 2019) to infer population dynamics and changes in demography over time. We used the ND2 dataset and performed four different runs: three of them based on geographic groups (Pacific, Chiapas and Yucatán_CA), and one based on the wholeA. rutila
complex. The Tres Marías group was not included in an independent run because of the lack of informative sites for ND2. We set a GTR + substitution model, a strict molecular clock, a Coalescent Bayesian Skyline as the tree prior, and a Piecewise-constant skyline model (Drummond et al. 2005) with five groups. We used a mutation rate of 0.0145 substitutions per site per lineage per million years for ND2, following Lerner et al. (2011), and ran each analysis through 20 million generations.Results
Phylogenetic analysis
The model of sequence evolution selected for the mtDNA data set was GTR + G (nst = 6, rates = gamma).The nucleotide composition of mtDNA was as follows:T = 23.9%, C = 34.5%, A = 30.8%, G = 10.6% for ND2; and T = 25.9%, C = 15.7%, A = 26.4%, G = 31.8% for COI. The parsimony informative sites for ND2 were 72 (605 bp),and 38 for COI (568 bp). For nuclear markers, the models of sequence evolution were: F81 (nst = 1) for MUSK,and GTR + I + G (nst = 6, rates = invgamma) for ODC.The nucleotide composition was: T = 29.6%, C = 19.4%,A = 29.4%, G = 21.4% for MUSK; and T = 36.4%,C = 17.3%, A = 24.7%, G = 21.4% for ODC. The parsimony informative sites for MUSK were 19 (628 bp), and 83 for ODC (571 bp).
According to the topology from the concatenated data set (mitochondrial and nuclear genes) under Bayesian Inference and Maximum Likelihood,A. rutila
forms a monophyletic group with respect toA. yucatanensis
andA. tzacatl
(see Fig. 2).
WithinA. rutila
, three major lineages can be identified: Pacific coast, Chiapas, and Yucatán Peninsula and Central America. There was also a well-supported clade (posterior probability = 0.93) within the Pacific phylogroup, which contained all individuals from the Tres Marías Islands. Within the Yucatán_CA phylogroup, four of the five individuals from Nicaragua grouped together into a well-supported clade (pp
= 0.99;bootstrap = 99). The individual phylogenies for mitochondrial loci (ND2 and COI) recovered three major lineages (Pacific, Chiapas, and Yucatán_CA) (see Additional file 2: Fig. S1). However, individual nuclear locus phylogenies did not recover any geographic structure (see Additional file 2: Fig. S1).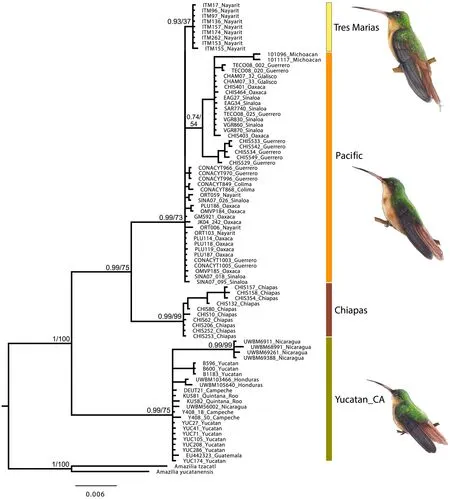
Fig. 2 Phylogenies with concatenated data (mtDNA: ND2, COI; nuclearDNA: MUSK, ODC) under Bayesian Inferences and Maximum Likelihood. Four main groups are represented in different colors (see geographic location in Fig. 1). Illustrations represent three different morphotypes based on differences in body size: Tres Marías–Islands (largest size, top), Pacific–continental (middle), Yucatán_CA (smallest size, bottom). Node supports are shown on main clades (posterior probability/bootstrap). Illustrations by Ana Hernández Ramírez
Genetic structure and neutrality tests
Deviations from neutrality were rejected for mitochondrial loci (ND2 and COI) and nuclear loci, except for the ODC locus (see Table 1). We found 21 haplotypes for ND2: 9 corresponded to the Pacific group, 9 to the Yucatán-Central America group, 2 to Chiapas, and 1 to the Tres Marías Islands. The Yucatán-Central America group was separated from the rest by 57 mutational steps(Fig. 1). For COI, we found 11 haplotypes in total: 4 corresponded to the Pacific group, 3 to Chiapas, 3 to Yucatán-Central America, and 1 to the Tres Marías Islands(shared with 2 individuals from the Pacific group from Guerrero). The Yucatán-Central America group wasseparated from the rest by 15 mutational steps (Fig. 1).The Pacific coast and Yucatán-Central America groups had the highest haplotype and nucleotide diversity (see Table 2).
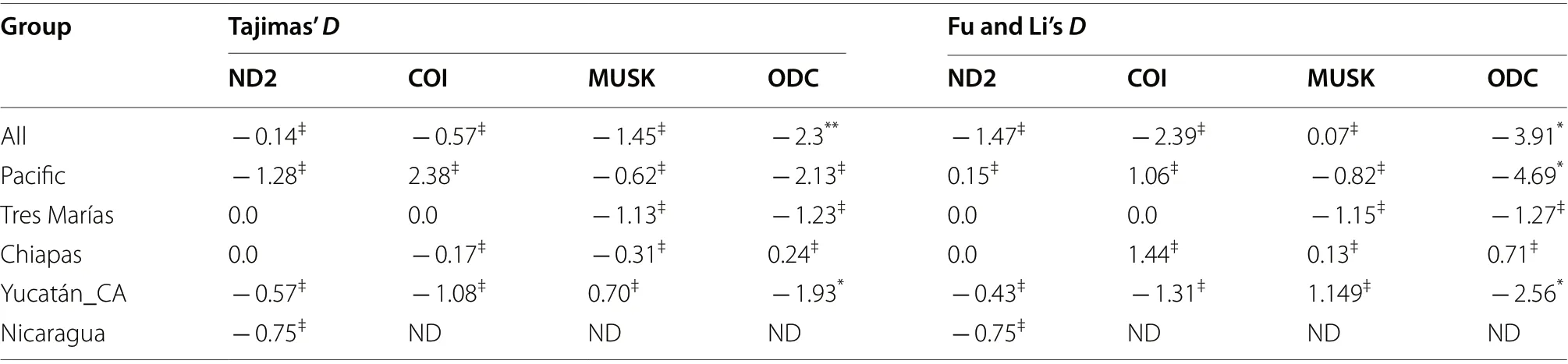
Table 1 Neutrality test by locus
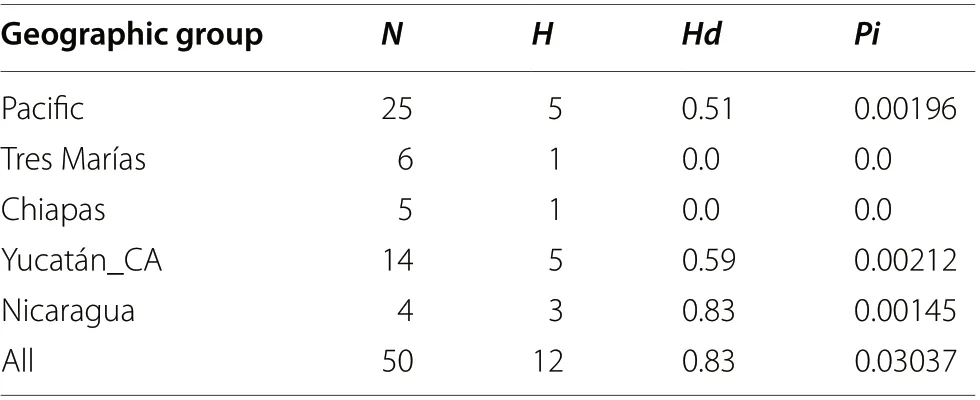
Table 2 mtDNA summary statistics
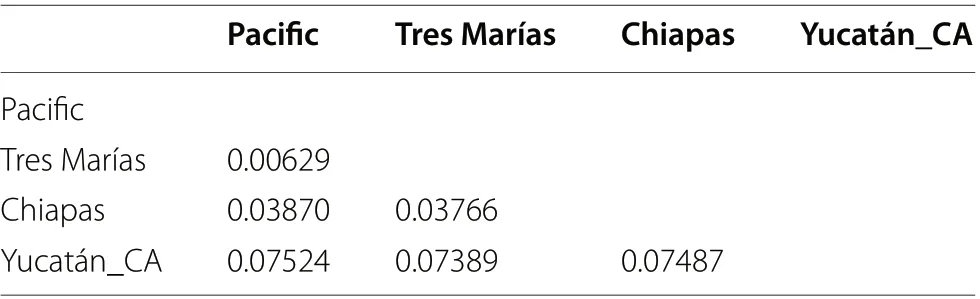
Table 3 mtDNA pairwise genetic distances among geographic groups in the Amazilia rutila complex
The mtDNA genetic distances among groups showed an overall genetic distance of 5.24% for the Pacific group,5.69% for Chiapas, and 7.52% for the Yucatán_CA group(Table 3). The pairwiseF
fixation indices were all significant and indicated strong population structure of geographic groups for mtDNA data (Table 4). We did not find strong differentiation between the Pacific and its subgroup from Guerrero. However, the Guerrero subgroup showed a more robust differentiation with respect to the Tres Marías group than to the rest of the Pacific subgroup. Substructure was detected within the Yucatán_CA group, with some Nicaraguan samples (UWBM-6911,UWBM-68991, UWBM-69261, and UWBM-69388) separated from the rest of the group, with anF
of 0.78549.AMOVA analyses revealed that most of the genetic variation was among geographic groups (96.26%), and there was little variation within populations (3.74%; Table 3).TheF
value from general AMOVA results indicated strong genetic structure and differentiation among populations (F
= 0.96; Table 5).The STRU CTU RE analyses detected two genetic groups (K
= 2; first-level analysis; See Additional file 3:Figs. S2, S3; Fig. 3). One group comprised all individuals from Sinaloa to Chiapas, including Tres Marías. The second group included the Yucatán Peninsula and Central American individuals. To evaluate potential substructure within these groups, we ran a hierarchically structuredanalysis. Individual structure analyses for the first group revealed two genetic groups (K
= 2), with no ancestral mixing between them. One group was composed of individuals from Chiapas and the second group recovered all of the Pacific clade including Tres Marías individuals.The substructure within the Yucatán_CA group recovered two groups (K
= 2). One group included samples from western Nicaragua and three samples from Yucatán. The second group included one sample from western Nicaragua, one sample from Guatemala and the rest of the Yucatán Peninsula individuals.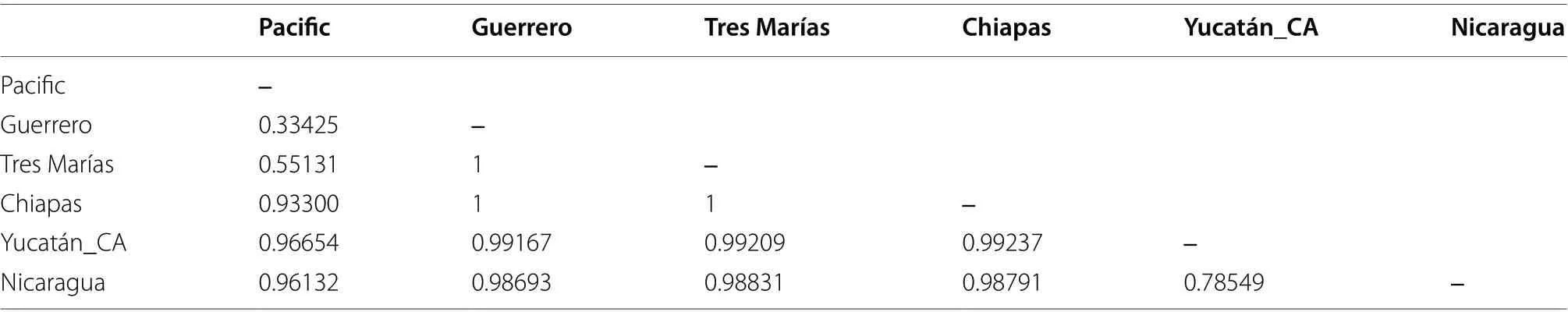
Table 4 Pairwise FST indices (below diagonal, *P ≤ 0.05) among populations in the Amazilia rutila complex
Divergence time estimates
Divergence times estimated that theA. rutila
complex split from its sister clade around 9.4 million years before present (Myr BP) (95% highest posterior density[HPD] 8.04‒10.87; Fig. 4), while theA. rutila
complex root was dated 7.05 Myr ago (95% HPD 5.88‒8.29).This estimate also corresponds to the split between the two main clades: (1) Pacific + Tres Marías and Chiapas, and (2) Yucatán_CA. The Pacific clade and Chiapas split around 3.99 Myr BP (95% HPD 3.16‒4.84).The Pacific Clade node origin was 1.65 Myr BP (95%HPD 0.81‒1.81 Ma). According to these estimates,A.rutila
arrived at the Tres Marías Islands 0.2 Myr BP(95% HPD 0.6‒0.44 Ma). The age of the continental subclades within the Pacific clade suggests that Pleistocene climatic changes may have caused contractions in the continental populations ofA. rutila
and resulted in incipient substructure, however although the TresMarías Islands is well supported, other clades are not supported. The Chiapas clade arose 0.76 Myr BP (95%HPD 0.43‒1.15 Ma).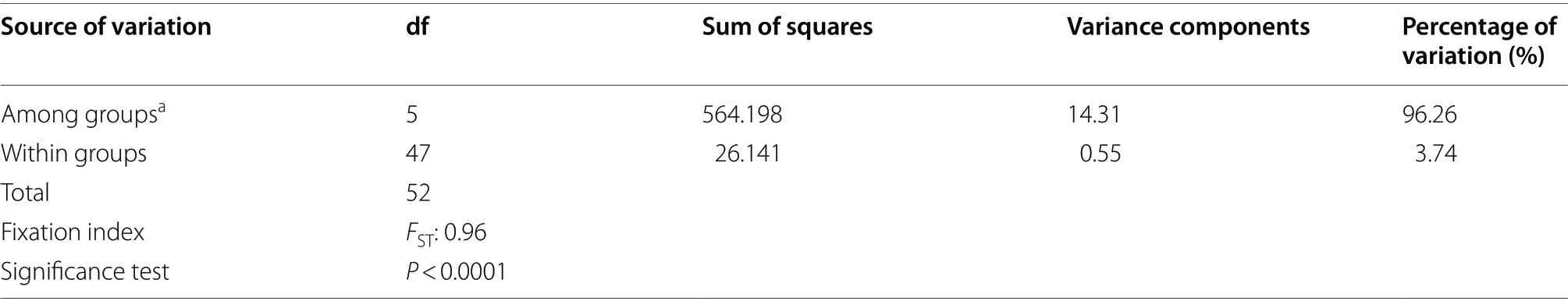
Table 5 mtDNA AMOVA comparing geographic groups in the Amazilia rutila complex
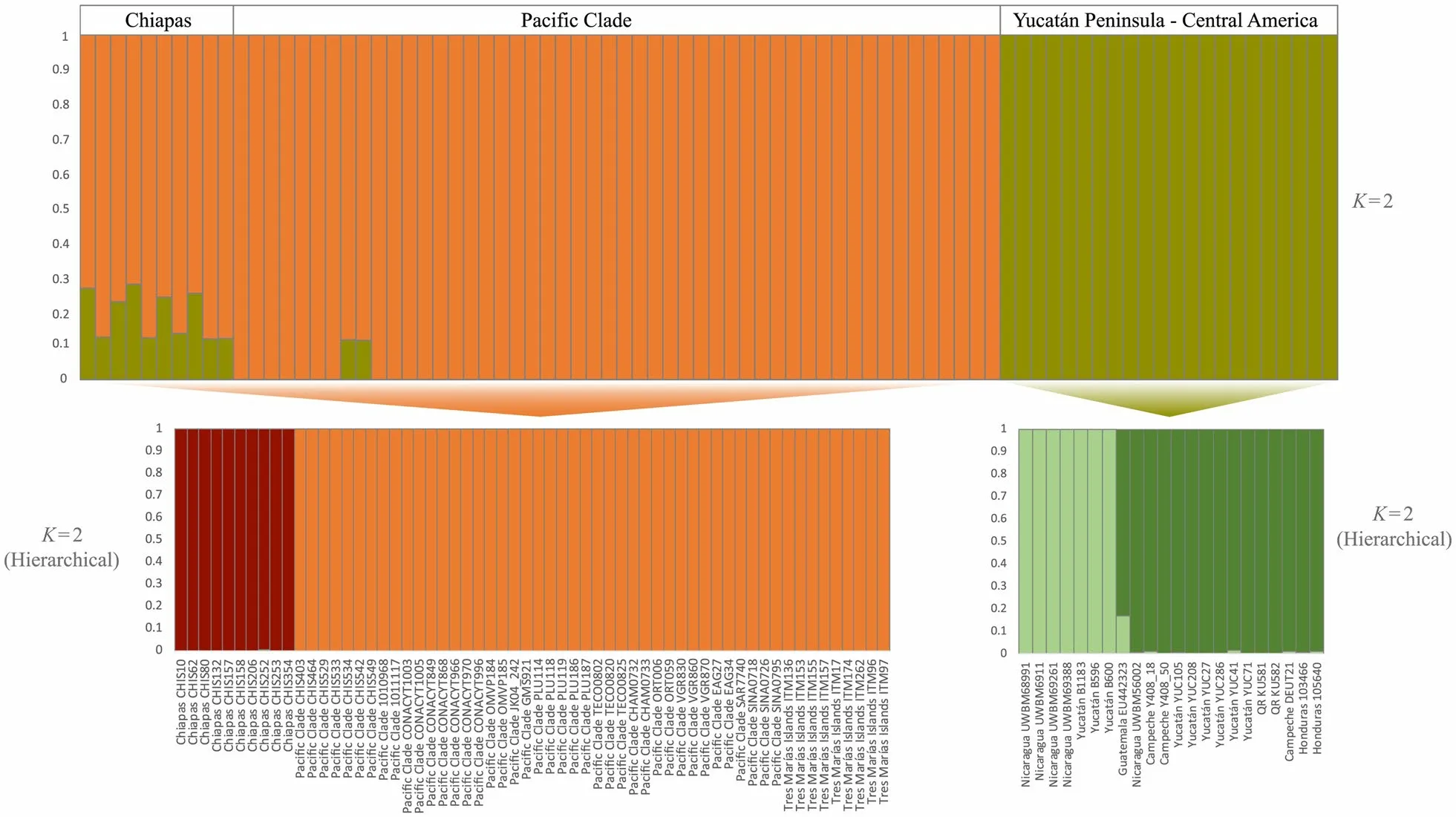
Fig. 3 STRU CTU RE results. The best fit K = 2 with the entire data set (above) and hierarchically within each genetic cluster (below: K = 2 for Pacific and Chiapas groups, and K = 2 for Yucatán_CA group)
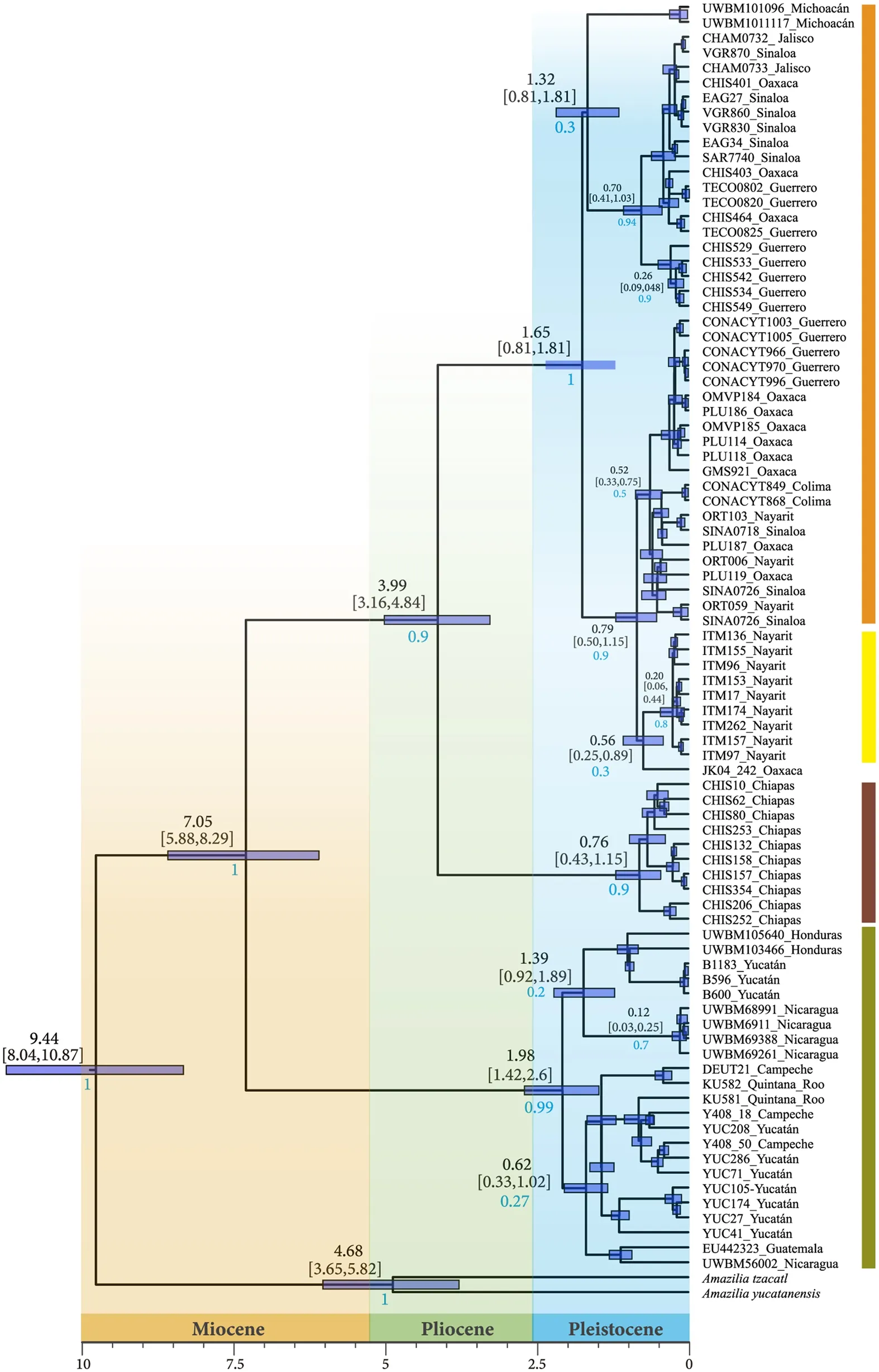
Fig. 4 Ultrametric phylogenetic tree obtained with Beast using concatenated matrix and different locus rates. Four main groups are represented with a vertical bar in different colors (see geographic location in Fig. 1). Posterior probabilities in blue
The crown node for the Yucatán_CA clade emerged 1.98 Myr BP (95% HPD 1.42‒2.6 Ma). Internal clades in the Yucatán_CA group were detected by Beast analysis, but they had weak node support, with the exception of the Nicaragua clade, which had intermediate support (0.7 posterior probability) and a date of 0.12 Myr BP (95% HPD 0.03‒0.25 Ma).
Morphometrics
We found minor sexual dimorphism in this species.Females were slightly but statistically significantly smaller than males in two body size variables we measured (WC,W
= 2196.5,p
< 0.001; TL,W
= 2194,p
< 0.001). However, the effect sizes were very small, so we carried out subsequent tests of geographic variation with both sexes pooled together.There were statistically significant differences among geographic groups (Kruskall-Wallis tests WC:χ= 78.86,p
< 0.001; TL: χ= 73.09,p
< 0.001; BH:χ= 47.45,p
< 0.001; CL: χ= 62.61,p
< 0.001; BW:χ= 90.30,p
< 0.001). The Tres Marías phylogroup had larger values than the three continental groups for all variables. This was consistent with the results of our PCA, which showed that the continental phylogroups largely overlapped in morphological measurements,but the Tres Marías group was significantly larger.Principal component 1 explained 74.75% of the cumulative variance and roughly corresponds to overall body size, with large contributions from Wing Chord(WC) and Tail Length (TL) (Fig. 5).Ecological Niche modelling onto past scenarios and demography
We obtained a total of 445 presence records forA.rutila
after filtering steps. The number of occurrence points for each group was: (1) Pacific: 144 records,(2) Chiapas: 20 records, and (3) Yucatán_CA: 281 records. The results of the PCA of climate variables for the whole complex showed that the first four principal components explained most of the variation(88%, see Additional file 4: Fig. S4). According to these results and correlation values, we selected the following bioclimatic variables: BIO5 (Max Temperature of Warmest Month), BIO7 (Temperature Annual Range),BIO11 (Mean Temperature of Coldest Quarter), BIO12(Annual Precipitation), BIO15 (Precipitation Seasonality), and BIO18 (Precipitation of Warmest Quarter).The average evaluation metrics for the model results were: training AUC = 0.7598 (St.Dv ± 0.005), and a threshold value of 0.4178 (Maximum training sensitivity plus specificity Logistic threshold). Projections onto past scenarios for the whole complex are shown in Additional file 5: Figs. S5, S6. The suitability of environmental conditions has changed somewhat over time, with subtle reductions from LIG to LGM and posterior arrangements after LGM through the MH period. Projections during LGM showed discontinuous distribution, which contrasts with the current continuous distribution for the species (Pacific coast and Central America). Independent analyses were run for each geographic group, in which the first two principal components explained most of the variation (see Additional file 5: Figs. S5, S6). The bioclimatic variables selected for the Pacific group were BIO1, BIO3,BIO4, BIO7, BIO13, BIO17 and BIO19; for the Chiapas group were BIO4, BIO5, BIO12, BIO13, BIO14, BIO15,BIO16, and BIO18; and for the Yucatán_CA group were BIO3, BIO4, BIO7, BIO11, and BIO13. The resulting metrics were as follows: Pacific AUC = 0.8046 (St.Dv ± 0.0473), threshold = 0.358; Chiapas AUC = 0.8184(St.Dv ± 0.1119), threshold = 0.4255; Yucatán_CA AUC = 0.6515 (St.Dv ± 0.0470), threshold = 0.4515.Projections for Pacific group predicted some areas where the Chiapas and Yucatán_CA groups are currently distributed (see Additional file 5: Figs. S5, S6).Also, for the projections in the Yucatán_CA group,suitable areas were predicted where the Pacific and Chiapas groups now occur. The most conservative projections for suitable habitats were recovered when the Chiapas group was modeled. Past projections revealed a reduction of suitability during LGM and an increase during MH periods in all cases. During the LIG period,the Pacific showed continuous suitable habitat along the Pacific coast to Central America. In LIG projections for Chiapas and Yucatán_CA, no suitable conditions were recovered (see Additional file 5: Figs. S5, S6).The BSP results showed that population dynamics and demography in theA. rutila
complex have been generally constant over time with a notable population reduction close to the present. Effective population size patterns showed a stationary trend in the geographic groups analyzed (Pacific, Chiapas, Yucatán_CA), with recent population reductions in the Pacific and Chiapas groups,and a reduction followed by a slight increase towards the present in the Yucatán_CA group (see Additional file 5:Figs. S5, S6).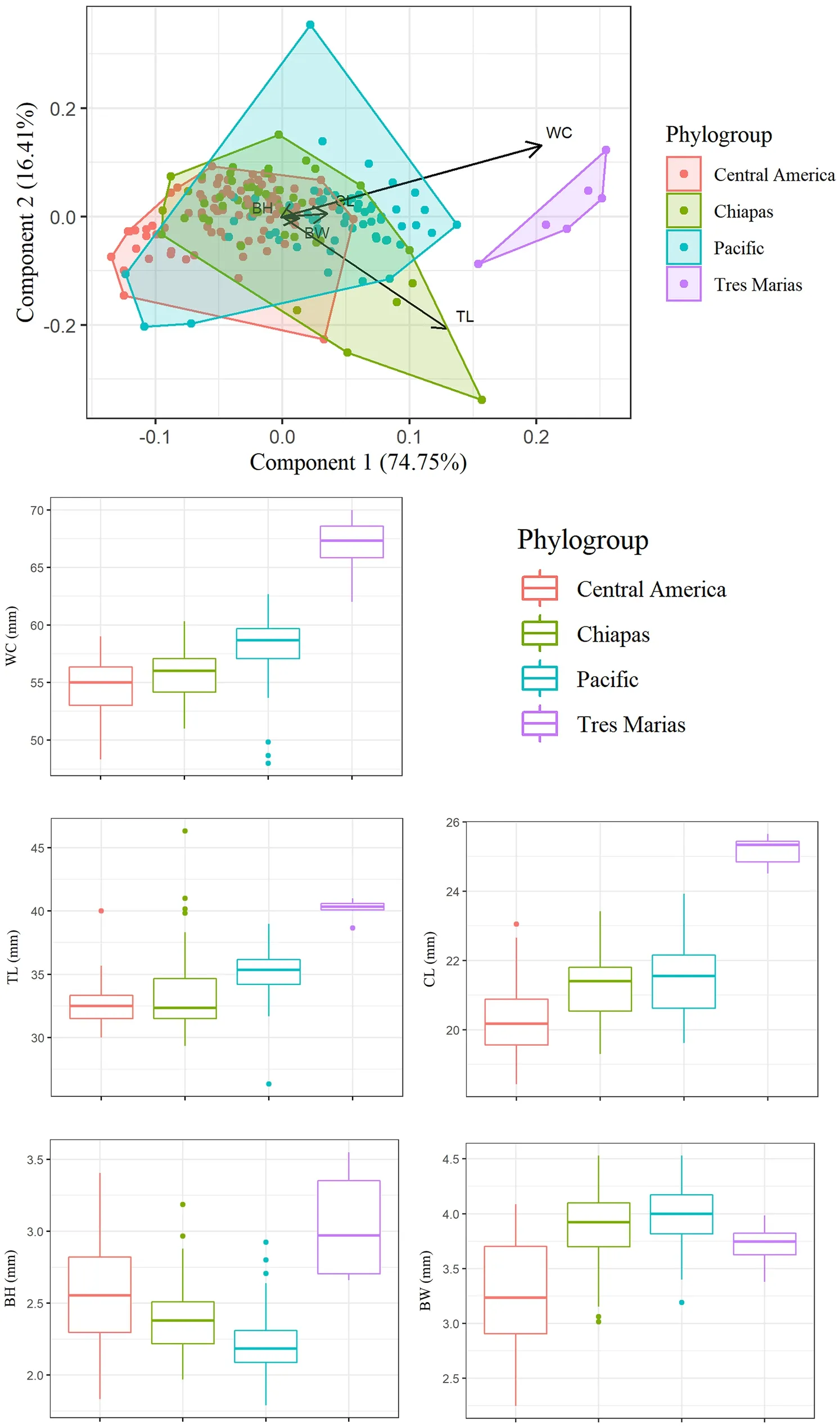
Fig. 5 Principal component analysis (PCA) of morphological data, showing the first two Principal Components, which explain 91.2% of the total variation (top); and box plots of each morphometric character measured. BW beak width, BH beak height, CL culmen length, TL tail length, WC wing chord
Discussion
Genetic variation and phylogeographic pattern
We found both deep and shallow genetic differentiation throughout the geographic distribution ofA. rutila
. The lineages corresponding to the Pacific clade (pp
= 0.99),Chiapas (pp
= 0.99) and the Yucatán Peninsula-Central America (pp
= 0.99) show deep structuring according to genetic distances,F
index, AMOVA, haplotype network, multilocus phylogeny and mtDNA phylogenies.Within the Pacific clade, we can distinguish clear structure for Tres Marías Islands individuals with a monophyletic clade (pp
= 0.94; geographically isolated),F
index,one haplotype for ND2 locus, and one haplotype for COI(shared with two samples from Guerrero); however, the structure analysis shows mixed ancestries between the Tres Marías Islands and Continental samples. Similarly,a multilocus phylogeny suggests a shallow substructure in the Yucatán Peninsula-Central America clade for West Nicaraguan samples (pp
= 0.99), which share ancestry with some samples from Yucatán.Despite minor differences in median-joining networks based on the ND2 versus COI markers (Fig. 1), the geographic groups had no shared haplotypes, with the exception of one haplotype that was shared between the Tres Marías group and three individuals from the Pacific group (COI marker). The information from nuclear genes that was included to construct the multilocus phylogenetic tree (see Additional file 2: Fig. S1) did not weaken the pattern found using mtDNA; the multilocus phylogeny maintained the same geographic structure with high node support values (posterior probabilities and bootstrap values), even of the retained ancestral polymorphisms, favored by nuclear signal and its reduced substitution rates compared to mtDNA (Moore 1995).A. rutila
belongs to the third youngest subclade of nine Trochilidae subfamilies, known as the Emeralds (Bleiweiss et al. 1997; McGuire et al. 2007, 2014). The tempo and mode of evolution in the hummingbird higher clades has been considered to be the product of a series of historical events including colonizations, extinctions, recolonizations, and the influence of mountain chain uplift,resulting in the radiation of highland species and the establishment of lowland taxa (Bleiweiss 1998; McGuire et al. 2014). Therefore, timing is crucial to understanding phylogeographic patterns inA. rutila.
This complex split from its sister group 7.05 Myr BP (95% HPD 5.88‒8.29 Myr BP), and the four main clades reported here arose 3.99‒0.20 Myr BP. During the late Miocene, Pliocene and Pleistocene, Middle American biological diversity was influenced by several major processes. One is the uplift of major mountain chains, which generated geographic barriers and created open niches in high-altitude and lowaltitude habitats that favored diversification (McCormack et al. 2008). Biodiversity was also influenced by the climatic changes of Pleistocene climatic oscillations, which created a mosaic of habitats that resulted in isolated populations.The Tres Marías phylogroup is embedded in the larger Pacific clade with a node origin dated 0.20 Myr BP (95%HPD 0.6‒0.44 Myr BP); this incomplete separation is a pattern that has been reported in other birds at early stages of speciation (e.g. Cortés-Rodríguez et al. 2008).STRU CTU RE analysis did not recover substructure for Tres Marías individuals or any Pacific samples. However,the ΔK
selection method has been previously shown to underestimate population structure (Janes et al. 2017).Results of the AMOVA andF
index suggest incipient population structure, and haplotype networks showed a clear differentiation at the population level. Additionally, the Tres Marías group was the most morphologically distinct compared to the other groups. The Pacific clade is represented as a polytomy in the resulting phylogenetic tree, and some clades are recovered within it but with no node support (see Additional file 2: Fig.S1). As mentioned, the single valid clade is composed of the Tres Marías Islands individuals. There are several examples of bird taxa in this area (The Pacific coast of Mexico and Tres Marías Islands) which show similar patterns of geographic variation and population structure (e.g.Habia rubica
: Ramírez-Barrera et al. 2018;Phaethornis longirostris‒P. mexicanus:
Arbeláez-Cortés and Navarro-Sigüenza 2013;Vireo hypochryseus
: Arbeláez-Cortés et al. 2014). Those studies found that the main forces acting as drivers of divergence were the presence of geographic and ecological barriers, as well as historical habitat fragmentation. This is not the case forA. rutila
along the Pacific coast, since no structured pattern was found. There are two possible explanations for this lack of geographic substructure. First, small sample sizes may cause some haplotypes to be missing, since we found many intermediate haplotypes. Another explanation is an incipient substructure produced by short periods of isolation during the Pleistocene. The age of the Pacific clade is 1.65 Myr BP (95% HPD 0.81‒1.81), and the age ranges estimated for subclades are 1.32 to 0.20 Myr BP,which corresponds with Pleistocene cycles. Thus, these mixed haplotypes could be the result of short periods of isolation during the LGM with secondary contact at the LIG and the present. This idea is supported by the LGM Pacific projections (see Additional file 5: Figs. S5, S6)and BSP analysis (Additional file 6: Fig. S7). In contrast,STRU CTU RE hierarchical analysis did not show different groups within the Pacific Clade (Fig. 2).The Chiapas clade is well defined, supported by genetic distance (5.69%) andF
values (0.97‒1.00). This region east of the Isthmus of Tehuantepec is mainly influenced by historical events between the middle and late Pliocene and marine transgressions and regressions during the late Pliocene (Barber and Klicka 2010; Peterson et al.2010). In general, the Isthmus of Tehuantepec represents a geographic barrier for highland bird species (e.g. Zamudio-Beltrán and Hernández-Baños 2018), but not for lowland species. However, some phylogeographic studies of lowland birds have described the influence of this low altitude zone (e.g.Campylorhynchus rufinucha
: Vázquez-Miranda et al. 2009). According to our time-calibrated tree, the Chiapas clade separated during Pleistocene climatic fluctuations ~ 760 kyr BP (95% HPD 0.43‒1.15 Ma),as there was a disruption of suitable conditions during the Last Glacial Maxima across the Isthmus of Tehuantepec(see Fig. 5). The Yucatán Peninsula and Central America clade also represents an independent entity, separated by 57 and 15 mutational steps, respectively, in the ND2 and COI trees (see Fig. 1), assuming low levels of gene flow,with an origin 1.98 Myr BP (95% HPD 1.42‒2.6 Myr BP).This genetic signal of isolation from the other groups may be influenced by the geographic distance and differences in environmental spaces. The Pacific and Chiapas group are distributed along the tropical deciduous forest belt, which has marked dry and rainy seasons and has been repeatedly identified as a distinct biogeographical province (i.e. Gordon and Ornelas 2000), while the Yucatán Peninsula has moderately pronounced dry and rainy seasons with relatively constant average temperature throughout the year (Paynter 1955). Also, our ENM past projections suggest periods of habitat contractions,during which populations from the Yucatán Peninsula and Central America could have been closer from each other (LGM MIROC, see Additional file 5: Figs. S5, S6),followed by periods of isolation. AlthoughA. rutila
is allopatrically distributed in the Yucatán Peninsula and Central America, the studied individuals were placed in a single group. This link between individuals close to the Yucatán Peninsula and Central America was also found inA. tzacatl
(Miller et al. 2011), in which populations in Tabasco (Mexico), Belize, Honduras, and Costa Rica shared several haplotypes despite the long distances between them, and the most differentiated populations were the southernmost populations in Panama. The periods of isolation mentioned above combined with demographic decreases (see BSP results) could explain the shallow substructure found in the West Nicaragua samples (pp
= 0.99). However, our STRU CTU RE analysis shows a mixed ancestry between those samples and some Yucatán individuals (Fig. 2).The influence of Pleistocene climatic fluctuations on processes of diversification and incipient speciation have been reported for many birds (e.g.Euphonia affinis
: Vázquez-López et al. 2020), including hummingbirds (e.g.Eugenes fulgens
: Zamudio-Beltrán et al. 2020).These habitat fluctuations are well documented as factors that promote population contractions, limiting gene flow (refugia hypothesis; Arango et al. 2021), as well as expansions, resulting in zones of contact between distant populations, with current signals of shared haplotypes and polyphyletic patterns (e.g. MesoamericanAmazilia
complex: Jiménez and Ornelas 2016; White-chestedAmazilia
complex: Rodríguez-Gómez and Ornelas 2018). Diversifications occurring throughout the Miocene, when the estimates of divergence dates of the genusAmazilia
are presumed ca. 19.85 Mya (i.e. Ornelas et al.2014), may have depended less on these climatic conditions. However, for events traced after this period, the rapid changes in climate cycling were crucial (Bleiweiss 1998). Furthermore, phylogeographic patterns of species sharing (totally or partially) the geographic distribution ofA. rutila
show similarities (e.g.Turdus rufopalliatus
:Montaño-Rendón et al. 2015; Ortíz-Ramírez et al. 2018;de Silva et al. 2020). In general, the diversification processes have resulted from a variety of events, sharing the explanations of an intricate orographic history, climatic fluctuations during the Pleistocene, and thus low signals of gene flow favored by the isolation of populations due to distance or historical refuges.Morphometrics
Our results showed little variation among the continental phylogroups of this species in the morphological characters we measured, despiteA. rutila
’s large geographic range. However, the Tres Marías Islands phylogroup is larger than the continental phylogroups in four out of five measurements (Fig. 3). This is consistent with several other avian species from Tres Marías (Grant 1965) and is a clear example of the “Island Rule” (van Valen 1973;Lomolino 1985; Clegg and Owens 2002; Boyer and Jetz 2010, Benítez-López et al. 2021). This shift in body size has been attributed to changes in thermal ecology (Clegg and Owens 2002), as the Tres Marías Islands are significantly drier and warmer than the continental habitat of the species. However, dramatic divergence in body size can be the result of geographic isolation independent of environmental pressure (Seeholzer and Brumfield 2018).This divergence in body size with relatively similar beak sizes suggest that beak morphology might be more tightly constrained than body size due to this species’ feeding ecology.Implications for taxonomy
We present a taxonomic proposal for theA. rutila
complex based on the recovery of four independent clades with high support values corresponding to natural geographic groups.Our results support the first differentiated clade,A. r.graysoni
, distributed in the Tres Marías Islands (Nayarit,Mexico). This differentiation was also confirmed by our morphometric analyses, which showed that the Tres Marías Islands population is the largest morphotype.A.r. graysoni
was first suggested as an independent species by Ridgway (1901), who describedAmazilia graysoni
(Grayson’s Hummingbird) as a separate species similar in coloration toA. rutila
, but darker and much larger.Then following the alternative phylogenetic/evolutionary species taxonomy of the Mexican avifauna, Navarro-Sigüenza and Peterson (2004) proposedA. r. graysoni
as an independent evolutionary species, which is morphologically distinct in size and coloration from the rest of the subspecies withinA. rutila
. The proposal of elevating this taxon to species level has been discussed recently(de Silva et al. 2020). The destruction of native habitat,the introduction of exotic species, and reduced population sizes increase the vulnerability of birds on the Pacific Islands of Mexico and must be immediately addressed in conservation policies (Hahn et al. 2012; de Silva et al.2020).The second differentiated clade corresponds to the group distributed along the Pacific coast. Within this second group, two subspecies have been described (A. r.rutila
andA. r. diluta
), which, according to our results,are not observable as separate groups. Therefore, we propose that the Pacific coast phylogroup be represented byA. r. rutila
(the first named).The third phylogroup is distributed in Chiapas, Mexico (east of the Isthmus of Tehuantepec). This group corresponds toA. r. corallirostris
, which is distributed from south and southeastern Mexico to Costa Rica. The Chiapas phylogrouop is distributed in an important and well-studied main fauna region, considered an area of endemism in western Mexico with one of the highest values of avian species richness (Peterson and Navarro 2000;García-Trejo and Navarro-Sigüenza 2004).Finally, the fourth independent group corresponds to the clade of the Yucatán Peninsula—Central America, the most genetically differentiated (7.52%), which also corresponds toA. r. corallirostris
. According to our results,A. r. corallirostris
is a polyphyletic subspecies, since it is composed of two independent groups.A. r. corallirostris
was described by Bourcier and Mulsant (1846) with specimens from Escuintla, Guatemala. Therefore, the nameA.corallirostris
should be applied to the Yucatán-CA clade(Ridgway 1901). This left the Chiapas group without a name, but specimens from Huehuetán, Chiapas were described underA. cinnamomea saturata
by Nelson in 1898, and this name was later considered a synonym ofA. r. corallirostris
(Ridgway 1901). As a consequence,the Chiapas lineage could be namedA. saturata
(Nelson 1898).Conclusions
The evolutionary hypothesis of diversification within this group is of geographic isolation (limited gene flow), differences in current environmental conditions, and historical habitat fragmentation promoted by past scenarios(Pleistocene refuges). TheA. rutila
complex consists of four well-defined clades that, in our opinion, merit taxonomic reevaluation. Our data suggest thatA. r. graysoni
(Tres Marías Islands) as well asA. r. rutila
(Pacific coast)should be considered full species. The other two strongly supported clades are: (a) the Chiapas group (southern Mexico), and (b) the populations from Yucatán Peninsula and Central America. These clades belong to theA. rutila corallirostris
taxon as currently defined. We propose that this group be split and renamed as two species:A. saturata
for the Chiapas group andA. corallirostris
for the Yucatán Peninsula and Central America group.Supplementary Information
The online version contains supplementary material available at https:// doi.org/ 10. 1186/ s40657- 021- 00295-0.
Additional file 1: Table S1. Information of voucher numbers (ID), geographic locations (longitude, latitude), and biological collections of tissue samples used in this study. Table S2. Information of accession numbers(AN) and geographic locations (locality, longitude, latitude) of GenBank’s sequences used in this study.
Additional file 2: Figure S1. The Bayesian Inference and Maximum Likelihood of the mitochondrial DNA and nuclear DNA.
Additional file 3: Figure S2. STRU CTU RE analysis ofAmazilia rutila
complex. Figure S3. Hierarchically structure analysis.Additional file 4: Figure S4. Statistics obtained for the selection of bioclimatic variables (19 variables from WorldClim) that were used on Ecological Niche Modelling projections, for the whole complex (Amazilia rutila
), and each geographic group (Pacific, Chiapas, Yucatán_CA).Additional file 5: Figure S5. Ecological Niche Modelling forAmazilia
rutila
under different past scenarios: LIG (Last Inter Glacial), LGM (Last Glacial Maxima), and MH (Mid Holocene) periods. Figure S6. Ecological Niche Modelling for each geographic group ofAmazilia rutila
under different past scenarios: LIG (Last Inter Glacial), LGM (Last Glacial Maxima), and MH(Mid Holocene) periods.Additional file 6: Figure S7. Bayesian skyline plots (BSP) for geographic groups (Pacific, Chiapas, Yucatán_CA), and for theA. rutila
complex.Acknowledgements
We thank M. Robbins of the University of Kansas (Natural History Museum),and S. Birks of the University of Washington (Burke Museum of Natural History and Culture) for providing tissue samples used in this study. We also thank Alejandro Gordillo Martínez, Fabiola Ramírez, Isabel Vargas-Fernandez and Raúl Ivan Martínez Becerril for technical support. We acknowledge the General Direction of Wildlife (Instituto de Ecología, SEMARNAT, México) for providing collecting permits. L. Kiere reviewed the English.
Authors’ contributions
BEH-B and MV-L designed the study. BEH-B secured financial support. MV-L carried out the laboratory work. MV-L, BEH-B, SMR-B and LEZ-B analyzed the data. BEH-B, MV-L, NC-R, AB-H, LEZ-B and KR contributed to the writing and improvement of the manuscript. All authors read and approved the final manuscript.
Funding
This research was supported by PAPIIT/DGAPA, Universidad Nacional Autónoma de México (UNAM) through a grant to Blanca E. Hernández-Baños(IN220620). LEZ-B acknowledges the Postdoctoral scholarship provided by DGAPA-UNAM.
Availability of data and materials
The sequences generated during the current study are available in GenBank(accession numbers: MZ998668-MZ998740, OK624605-OK624657, OK614044-OK614080, OK618334-OK618361).
Declarations
Ethics approval and consent to participate
This study was performed in line with the principles of the Declaration of Helsinki.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Author details
Museo de Zoología, Facultad de Ciencias, Universidad Nacional Autónoma de México, Apartado Postal, 04510 Mexico City, Mexico.Department of Biology, Ithaca College, Ithaca, NY 14850, USA.Museo de Zoología, Facultad de Estudios Superiores Zaragoza, Universidad Nacional Autónoma de México,Mexico City, Mexico.Department of Biology, Colorado State University, Fort Collins, CO 80523, USA.
Received: 19 February 2021 Accepted: 31 October 2021

杂志排行

Avian Research的其它文章
- Taxonomic revision of the Savanna Nightjar (Caprimulgus affinis) complex based on vocalizations reveals three species
- Taxonomic status of grey-headed Yellow Wagtails breeding in western China
- The composition of mixed-species flocks of birds in and around Chitwan National Park,Nepal
- True grit: ingestion of small stone particles by hummingbirds in West Mexico
- Stopover behavior of Red-eyed Vireos (Vireo olivaceus) during fall migration on the coast of the Yucatan Peninsula
- Plastering mud around the entrance hole affects the estimation of threat levels from nest predators in Eurasian Nuthatches