肽钙螯合物LQVLEK-Ca的制备及表征分析
2022-01-05郑金玲孙小东庄永亮
樊 建,郑金玲,孙小东,庄永亮
(昆明理工大学 农业与食品学院,云南 昆明 650500)
0 引 言
钙是人体内重要的无机元素,参与了许多生理过程,从营养健康到生长发育起着关键性作用,人体内钙的缺乏会导致许多器官和功能的疾病[1-2].研究发现,生物活性肽能够与钙离子结合,形成稳定的肽钙螯合物,肽钙螯合物具有自身的吸收转运机制[3].肽钙螯合物既可以将钙元素以螯合物的形式被完全吸收,又能有效地释放钙离子,具有钙吸收率好、生物利用率高等特点[4].因此,利用生物活性肽与钙离子的螯合特性,制备肽钙螯合物促进钙的吸收,从而提高补钙效果.
本文利用LQVLEK与无机钙进行螯合反应制备肽钙螯合物LQVLEK-Ca,通过紫外光谱(UV-Vis)、扫描电子显微镜(SEM)和X射线衍射(XRD)等技术方法对LQVLEK-Ca进行结构表征.利用傅里叶变换红外光谱(FTIR)和超高效液相色谱-四级杆-轨道阱-质谱联用(UPLC-Q-Orbitrap-MS2)进一步鉴定肽钙螯合物的结合位点,并采用软件模拟方法,预测LQVLEK-Ca的分子结构.
1 材料和方法
1.1 材料试剂
LQVLEK由上海杰肽生物科技有限公司提供.色谱级乙腈和甲酸购自德国 Merck KgaA 公司.优级纯甲醇购买于阿拉丁生化科技有限公司.其它试剂均为分析纯.
1.2 仪器与设备
novAA®350 原子吸收仪 德国 Analytikjena 公司
TU-1901 紫外可见分光光度计 北京普析通用仪器有限公司
Tecnai G2 TF30 扫描电子显微镜 美国 Thermo Scientific 公司
X 射线衍射仪 荷兰 PANalytical B.V.公司
Tensor 27 傅立叶变换红外光谱仪 德国 Bruker Optics 公司
超高效液相色谱-四级杆-轨道阱-质谱联用仪 美国 Thermo Scientific 公司
1.3 实验方法
1.3.1 肽钙螯合物的制备
称取LQVLEK 1 mg溶于1 mL去离子水中,添加2 mL 5 mmol/L CaCl2溶液.将混合液的 pH 调节至 7.8,在 37 ℃条件下恒温水浴 30 min.向混合液中加入4 mL磷酸钠缓冲液(20 mmol/L,pH 7.8),在 37 ℃下恒温水浴 30 min.将混合液离心(5 000 r/min,10 min),收集上清液,浓缩冻干,制备肽钙螯合物LQVLEK-Ca[5].
1.3.2 钙螯合能力测定
称取LQVLEK-Ca 100 mg,采用火焰原子吸收光谱法(FAAS),参照 GB 5009.92—016 中的方法测定LQVLEK-Ca中的钙含量,计算LQVLEK的钙螯合能力.
1.3.3 肽钙螯合物的结构鉴定
1)UV-Vis 分析
分别将LQVLEK和LQVLEK-Ca溶于去离子水中至相同的肽浓度.用去离子水调零后,在190 nm至400 nm 范围内进行扫描,扫描速度为2 nm/s,分辨率为1 nm.
2)SEM分析
分别取LQVLEK和LQVLEK-Ca 1 mg,均匀地涂在粘有双面导电胶带的标本架上,喷金镀膜,使用扫描电子显微镜观察并拍照.分析条件为:加速电压为15.0 kV,工作距离为12.8 mm,放大倍数为20 000×,工作温度为25 ℃.
3)XRD 分析
分别取LQVLEK和LQVLEK-Ca 10 mg,均匀研磨,使用XRD衍射仪收集数据.分析条件为:扫描角为2θ,范围为5°至90°,扫描速度为4°/min.
4)FTIR 分析
分别取LQVLEK和LQVLEK-Ca 1 mg,与100 mg干燥的 KBr 混合,均匀研磨,压片,使用FTIR在4 000至500 cm-1范围内进行扫描,光谱分辨率为4 cm-1.
5)UPLC-Q-Orbitrap-MS2分析
(1) 样品处理:将LQVLEK和LQVLEK-Ca溶于超纯水中,浓度为2 mg/mL,过0.22 μm的滤膜备用.
(2) 色谱条件:Hypersil Gold C18(1.9 μm,2.1 mm×100 mm);流动相 A: 0.1%甲酸-乙腈;流动相B:0.1%甲酸-水;进样量:2 μL;流速:0.2 mL/min;柱温:30 ℃;洗脱梯度:0~1 min(5% A 平衡),1~2.5 min(5%~10% A),2.5~12.5 min(10.0%~25.0% A),12.5~20 min(25.0%~52.5% A),20~22 min(52.5%~95.0% A),22~24 min(95.0% A),24~25 min(95.0%~5.0% A),25~30 min(5.0% A).
(3) 质谱条件: 扫描方式:Full MS,dd-MS2;分辨率:Full MS 35 000, dd-MS217 500;扫描范围:200~2 000 m/z;扫描离子:正离子模式;碰撞能量:10 eV,20 eV,30 eV.
6)LQVLEK-Ca结构预测
通过MOE(Molecular Operating Environment)软件中的Conformation Search模块,采用LowModeMD方法对LQVLEK和钙离子分子动力学构象进行搜索.设定最高能量为10 kcal,最大搜索构象数目为10 000.搜索获取较为稳定的构象结果由MOE软件记录,原子间的相互距离由MOE中的工具计算并标出.
1.4 统计分析
每个实验重复3次,数据表达为平均值±标准偏差.采用Origin 8.5软件进行数据处理和作图.
2 结果与讨论
2.1 肽钙螯合能力
结果表明,LQVLEK的钙螯合能力为 53.77±2.87 μg/mg,经计算,LQVLEK-Ca中肽和钙的摩尔比约为1∶1.与前期研究的生物活性肽的钙螯合能力相当[6-7],表明LQVLEK对钙离子具有较高的螯合能力.
2.2 肽钙螯合物的结构表征
2.2.1 UV-Vis分析
紫外光谱是评价分子结构表征的有效方法之一,通过紫外吸收光谱的强度和位移的变化可以判断生物活性肽与金属离子之间的结合情况[8].如图1所示,LQVLEK在 200 nm 附近显示出高强度的吸收峰,这是对应肽链中酰胺键C=O的n→π*跃迁产生的特征峰.与钙离子螯合后,LQVLEK-Ca可以观察到吸收峰强度明显增加,这可能是由于LQVLEK螯合钙后,导致酰胺键的电子云和配体的吸收特性发生变化而引起的.Zhao等[9]研究发现,二肽螯合物Gly-Tyr-Ca中酰胺键的吸收峰强度高于二肽Gly-Tyr.UV-Vis结果表明,LQVLEK和钙离子之间发生了螯合反应,证实了LQVLEK-Ca是新的化合物.
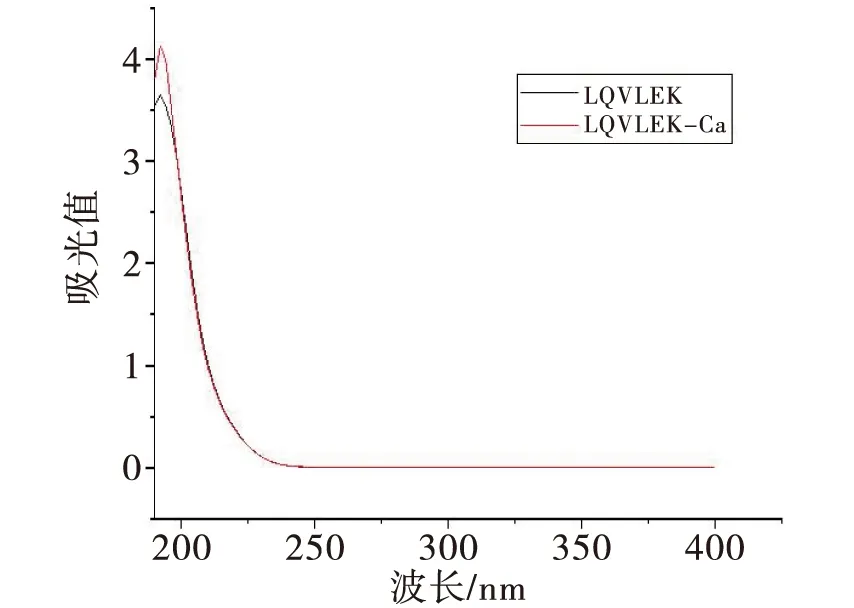
图1 LQVLEK和LQVLEK-Ca的紫外光谱图Fig.1 UV-Vis spectra of LQVLEK and LQVLEK-Ca
2.2.2 SEM分析
LQVLEK和LQVLEK-Ca的表面微观结构如图2所示.从图2(A)可以看出,LQVLEK的表面光滑,呈片状或块状,孔隙大而疏松.图2(B)显示LQVLEK-Ca的微观表面与LQVLEK有明显不同.LQVLEK在螯合钙离子后,光滑且疏松多孔的结构变成了排列致密的颗粒结构,且密度变大,这可能是LQVLEK与钙离子结合形成螯合物所致[10].此外,在LQVLEK-Ca表面还观察到一些类似晶体的结构,这可能是由于钙盐晶体吸附在肽表面所致[11].SEM结果表明,LQVLEK与钙离子之间存在相互作用,形成了一种新的肽钙螯合物.

(A) (B)图2 LQVLEK(A)和LQVLEK-Ca(B)的扫描电镜图像Fig.2 SEM of LQVLEK (A) and LQVLEK-Ca (B)
2.2.3 XRD分析
X射线衍射是一种通过结晶变化研究物质微观结构的分析方法[12].图3(A)显示LQVLEK的衍射图谱,在2θ为8.3°和18.9°附近有两个主要的衍射峰,LQVLEK没有尖锐的衍射峰,只有宽分散和弱分散的衍射峰,表明LQVLEK在结构上并没有形成有序排列,为无规则的非晶型结构[13-14].如图3(B)所示,LQVLEK与钙离子结合后,原有在2θ为8.3°附近的衍射峰消失,18.9°附近的衍射峰移至23.1°附近,并且在31.7°和45.4°出现强而尖锐的新的衍射峰,这表明LQVLEK与钙离子结合后,导致散射强度的急剧增加.XRD结果表明,LQVLEK与钙离子发生螯合反应,形成了新的晶体结构.
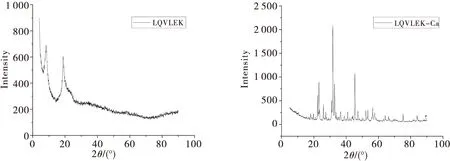
(A) (B)图3 LQVLEK(A)和LQVLEK-Ca(B)的X射线衍射图Fig.3 XRD spectra of LQVLEK (A) and LQVLEK-Ca (B)
2.2.4 FTIR分析
红外光谱常常用于分析有机官能团的形成和组成情况,主要包括O-H、C-O和N-H等.肽和肽钙螯合物的红外特征吸收峰的变化可以反映肽与钙形成相互作用的配体基团[15].LQVLEK和LQVLEK-Ca的FTIR光谱如图4所示.LQVLEK在3 431.14 cm-1和3 266.46 cm-1处有高频吸收,是由 N-H的伸缩振动引起的[16].LQVLEK与钙离子结合后,N-H振动移至3 449.76 cm-1和3 406.8 cm-1,这可能是N-H的电子云密度由于感应效应或偶极场效应变强引起的[17].该结果表明肽链中的氨基基团参与了与钙离子的螯合反应.LQVLEK在1 632.51 cm-1观察到由C=O键伸缩振动引起的酰胺-I振动,以及在1 543.73 cm-1处发现由C-N键伸缩振动引起的酰胺II振动.结合钙离子后,LQVLEK-Ca在酰胺-I带吸收峰移至1 633.95 cm-1处,酰胺II带的吸收峰移至1 555.18 cm-1处,这表明C=O和C-N可能参与了与钙离子的结合.先前研究表明,羰基氧存在的非键自由电子对可能有助于钙离子的螯合[17],这与本研究结果相似.
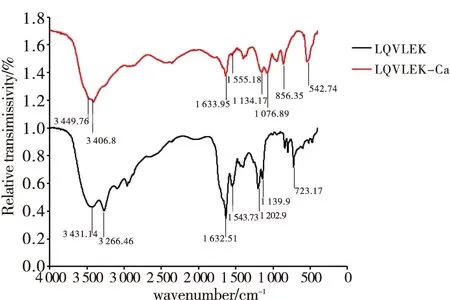
图4 LQVLEK(A)和LQVLEK-Ca(B)的红外光谱图Fig.4 FTIR spectra of LQVLEK (A) and LQVLEK-Ca (B)
在指纹区,LQVLEK在1 202.9 cm-1和1 139.9 cm-1处的吸收峰在螯合钙后分别移至1 134.17 cm-1和1 076.89 cm-1处,此类吸收峰的偏移可能是由于羰基氧和钙离子被未束缚的自由电子螯合而形成-COOCa导致的[18].此外,在1 000~500 cm-1之间的多个吸收峰中出现了变化,这可能是由C-H键和N-H键弯曲振动变化引起的.FTIR结果表明,LQVLEK-Ca是不同于肽的一种新的化合物,证实了LQVLEK与钙离子之间发生了螯合反应,并且螯合反应可能与羧基上的氧原子和氨基上的氮原子有关.
2.2.5 UPLC-Q-Orbitrap-MS2分析
质谱技术可以获取肽钙螯合物中螯合位点的信息,是肽钙螯合物结构解析的有效方法[7].LQVLEK和LQVLEK-Ca的质谱结果如图5所示.从图5(A)可以看出,m/z为365.72 Da代表的是[M+2H]2+分子离子峰,m/z为730.43 Da代表的是[M+H]+双电荷分子离子峰.图5(B)为LQVLEK的二级质谱图,b型碎片代表N末端片段,y型片段代表C末端片段[19-20].LQVLEK-Ca的一级质谱和二级质谱如图5(C)和5(D)所示.LQVLEK-Ca的一级质谱图显示m/z为265.20 Da、384.69 Da和751.38 Da的信号峰分别对应 [M+Ca+H]3+、[M+Ca]2+和[M+Ca-OH]+的分子离子峰.选择m/z为265.2的离子[M+Ca+H]3+进行二级质谱分析,如图5(D)所示.LQVLEK-Ca裂解为b型碎片和y型碎片,m/z为252.16、349.25、364.24和588.32处的碎片离子信号峰分别代表[b2+Ca-H]+、[b3+Ca-OH]+、[b3+Ca-2H]+和[b5+Ca-2H2O]+的峰值,这表明钙离子的结合位点可能位于LQ、LQV和LQVLE片段的N末端,并且LQ片段存在一个钙离子结合位点[21].m/z为314.16和444.20处的两个碎片分别对应[y2+Ca-2H]+和[y3+Ca-H]+,表明与钙离子结合位点可能位于EK和LEK片段的C末端,钙离子的结合位点可能存在于Lys中羧基的氧原子.
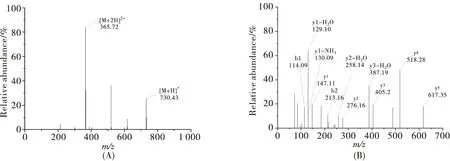

图5 LQVLEK与LQVLEK -Ca的质谱图:(A)LQVLEK的一级质谱图;(B)LQVLEK 的二级质谱图;(C)LQVLEK-Ca 的一级质谱图;(D)LQVLEK -Ca 的二级质谱图Fig.5 Mass spectrum of LQVLEK and LQVLEK-Ca: (A) primary mass spectrometry of LQVLEK; (B) secondary mass spectrometry of LQVLEK; (C) primary mass spectrum of LQVLEK -Ca; (D) secondary mass spectrum of LQVLEK -Ca
2.3 肽钙螯合物的分子结构预测
对比FTIR和UPLC-Q-Orbitrap-MS2结果发现,LQVLEK在结合钙离子的过程中,-NH2和-COOH发生了明显的变化,说明氨基上的氮原子和羧基上的氧原子可能参与了螯合反应.LQVLEK与钙离子的结合位点可能主要来自于位于端肽的氨基氮原子和羧基氧原子.采用MOE软件推测LQVLEK-Ca的分子结构如图 6 所示.氧原子在LQVLEK螯合钙离子作用中有明显的贡献,且LQVLEK-Ca整体形成了环状螯合结构.先前的研究表明,由于肽链长短和空间结构不同,存在空间位阻情况也不相同,钙离子的结合位点也有差异,但一般情况下钙离子与氨基酸末端氨基和羧基较容易发生螯合反应形成环状螯合结构[22-23],这与我们的结果一致.
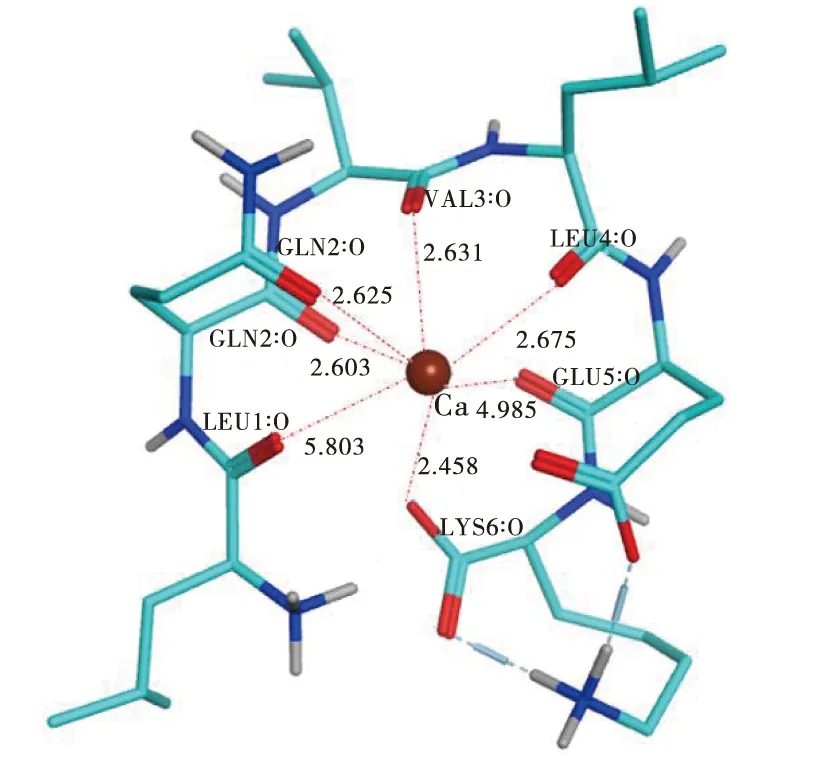
图6 LQVLEK-Ca分子结构图的预测Fig.6 The predicted molecular structure of LQVLEK-Ca
3 结 论
利用LQVLEK与氯化钙制备肽钙螯合物LQVLEK-Ca,LQVLEK和钙的螯合摩尔比约为1∶1.通过UV-Vis,SEM,XRD,FTIR 和 UPLC-Q-Orbitrap-MS2等技术手段对LQVLEK和LQVLEK-Ca的结构进行了比较分析,发现LQVLEK-Ca为一种新的化合物,其分子结构可能为环状螯合结构.本文研究可以为肽钙螯合物的制备、结构分析和实际应用提供一定的理论基础.
