基于指纹图谱及一测多评法的芍药甘草汤 物质基准质量评价研究
2021-12-30刘紫璇吴样明蒋鑫铖季巧遇李诒光江西中医药大学药学院南昌330004江中药业股份有限公司南昌330004
刘紫璇,吴样明,蒋鑫铖,季巧遇,*,李诒光,(.江西中医药大学药学院,南昌 330004;.江中药业股份有限公司,南昌 330004)
芍药甘草汤收录于国家中医药管理局会同国家药品监督管理局制定并公布《古代经典名方目录(第一批)》[1]。芍药甘草汤出自汉代张仲景所著《伤寒论》,可养阴柔肝、缓急止痛。现代药理研究表明,芍药甘草汤多以加减方应用于治疗解痉抗炎镇痛[2]。国家药品监督管理局发布《古代经典名方中药复方制剂简化注册审批管理规定》在借鉴日本汉方药管理经验的基础上,引入了物质基准的管理要求,以其作为质量控制的基准[3]。
在中药制剂质量控制方面,关键是有效性和安全性。由于中药作用的整体性,中药成分和作用机制的复杂性,多指标质量评价已成为行业的共识。目前,一测多评(QAMS)法多应用于中药材及中药复方制剂的质量评价[4-6]。本实验采用QAMS法,以芍药苷作为参照物,建立其他已知成分相对于参照物的相对校正因子,可同时测定8种成分的含量。建立了芍药甘草汤物质基准指纹图谱并进行相似度评价,以分析不同批次间样品的质量差异,为芍药甘草汤的质量评价提供参考。
1 材料
1.1 仪器
Thermo Ultimate 3000高效液相色谱仪(美国Thermo Fisher Scientific公司);Agilent 1260 Infinity 型高效液相色谱仪(美国Agilent公司);Milli-Q IQ 7000超纯水仪(美国Millipore公司);MSA224S-1CE-DA型万分之一分析天平、CPA225D型十万分之一分析天平、MSA6.6S-OCE-DM型百万分之一分析天平(德国sartorius 公司);SB-500DTY型扫频超声波清洗机(宁波新芝生物科技有限公司)。
1.2 试药
芍药苷(批号:110736-202044,纯度:96.8%)、甘草苷(批号:111610-201908,纯度:95%)、甘草酸铵(批号:110731-202021,纯度:96.2%)对照品(中国食品药品检定研究院);芍药内酯苷(批号:19121705,纯度:99.28%)、芹糖甘草苷(批号:20081705,纯度:98%)、异甘草苷(批号:17042102,纯度:98%)对照品(成都普菲德公司);苯甲酰芍药苷(批号:ST05580220,纯度:95%)、甘草素(批号:ST07000120,纯度:98%)对照品(上海Standard公司);乙腈为色谱纯,甲醇、磷酸为分析纯,水为超纯水。经考证,芍药甘草汤中所用白芍和甘草为毛茛科植物芍药Peaonia lactifloraPall.的干燥根和豆科植物甘草Glycyrrhiza uralensisFisch.的干燥根和根茎,其中甘草的炮制方法为清炒[7-10]。15批芍药甘草汤物质基准所用白芍和甘草药材经江中药业股份有限公司鉴定人员谢斌鉴定为毛茛科植物芍药Peaonia lactifloraPall.的干燥根和豆科植物甘草Glycyrrhiza uralensisFisch.的干燥根和根茎,由江中饮片厂炮制。饮片信息见表1。
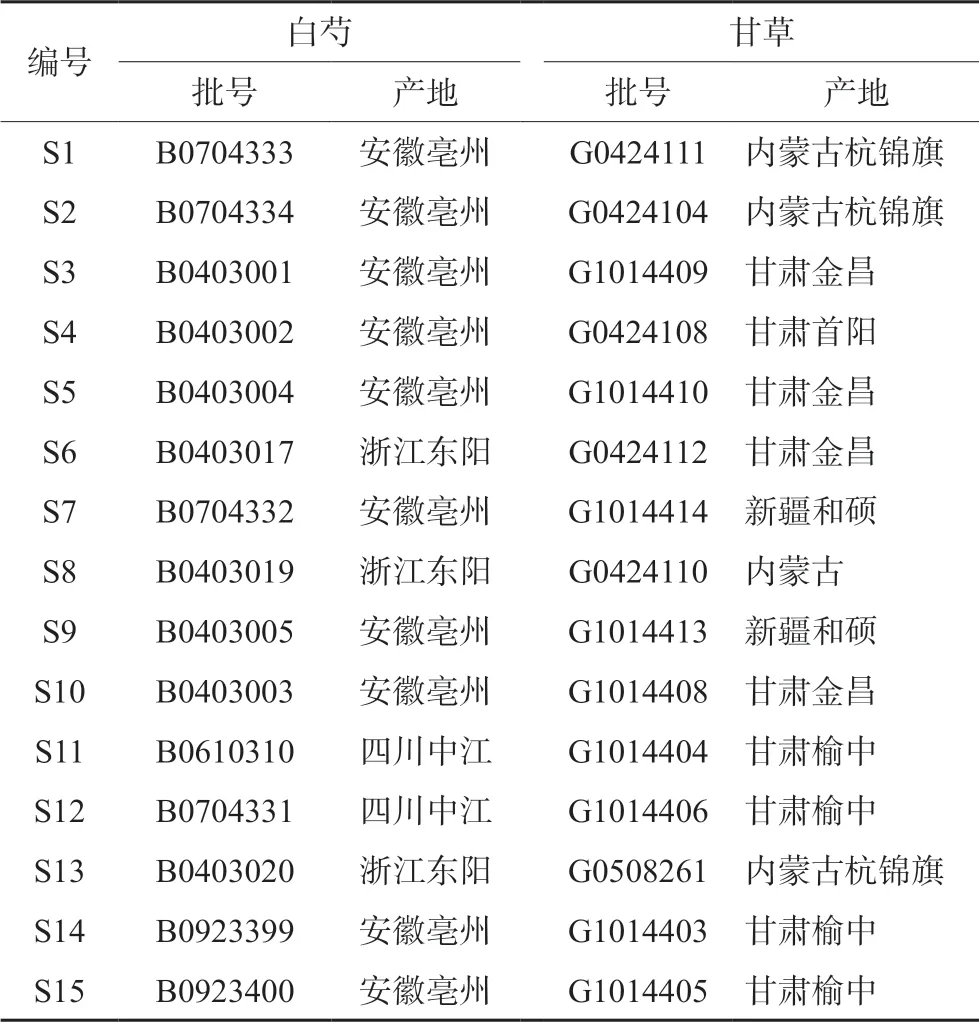
表1 样品信息 Tab 1 Information of sample
2 方法与结果
2.1 色谱条件
色谱柱为Agilent ZORBAX SB-C18(250 mm× 4.6 mm,5 μm);流动相为乙腈(A)-0.1%磷酸水溶液(B),梯度洗脱(0~10 min,10%~ 20%A;10~22 min,20%A;22~35 min,20%~40%A;35~50 min,40%A);检测波长230 nm;流速1.0 mL·min-1;柱温30℃;进样量10 μL。
2.2 混合对照品溶液的制备
分别精密称取芍药内酯苷、芍药苷、芹糖甘草苷、甘草苷、异甘草苷、甘草素、苯甲酰芍药苷、甘草酸铵对照品适量,精密称定,加70%甲醇配制成质量浓度分别为40.457、223.751、18.953、83.866、16.299、2.094、7.783、158.763 mg·mL-1的混合对照品溶液。
2.3 供试品溶液的制备
2.3.1 芍药甘草汤物质基准的制备 芍药甘草汤记载于汉代张仲景所著的《伤寒杂病论》,“白芍药、甘草各四两(炙)。上二味,以水三升,煮取一升五合,去滓,分温再服。”取白芍饮片、甘草饮片各约 55.2 g,加水 600 mL 浸泡 30 min,加热至沸腾,保持沸腾,煮至 300 mL,趁热过滤,减压浓缩,冻干,即得芍药甘草汤物质基准冻干粉。缺白芍样品和缺炙甘草样品的冻干粉,按上述方法制备。
2.3.2 供试品溶液的制备 取芍药甘草汤物质基准冻干粉适量,研细,取约0.1 g,精密称定,置于100 mL量瓶中,加入70%甲醇适量,超声(600 W,40 Hz)30 min,放冷,定容,摇匀,滤过,取续滤液,即得。
2.4 方法学验证
2.4.1 线性关系考察 将“2.2”项下混合对照品溶液按对半稀释法稀释4次,得到不同质量浓度的混合对照品溶液,按“2.1”项下色谱条件进行分析,以质量浓度(X,μg·mL-1)为横坐标,峰面积(Y)为纵坐标,绘制标准曲线,得到线性回归方程,结果见表2。
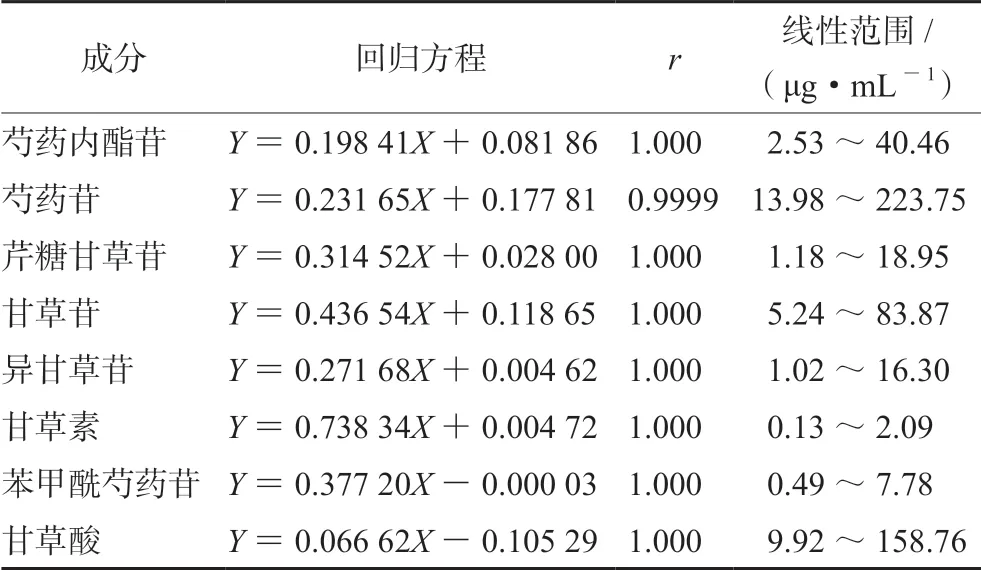
表2 各成分的线性关系考察结果 Tab 2 Linear regression of 8 components
2.4.2 精密度试验 精密吸取混合对照品溶液,按“2.1”项下色谱条件连续进样测定6次,记录峰面积和保留时间,结果7种成分相对于芍药苷的相对保留时间RSD均小于0.5%,8种成分峰面积RSD均小于0.96%,表明仪器精密度良好。
2.4.3 重复性试验 平行制备6份供试品溶液,按“2.1”项下色谱条件进样测定,记录峰面积和保留时间,结果7种成分相对于芍药苷的相对保留时间RSD均小于0.13%;芍药内酯苷、芍药苷、芹糖甘草苷、甘草苷、异甘草苷、甘草素、苯甲酰芍药苷、甘草酸的平均含量分别为1.15%、4.47%、0.55%、1.96%、0.37%、0.06%、0.18%、3.62%,RSD分别为0.61%、0.53%、1.3%、0.67%、1.0%、1.1%、0.59%、0.53%,表明本方法重复性良好。
2.4.4 稳定性试验 取同一批供试品溶液,分别于0、2、4、8、10、12、24、48 h按“2.1”项下色谱条件进样测定,记录峰面积和保留时间,计算得到结果7种成分相对于芍药苷的相对保留时间RSD均小于0.11%,相对峰面积RSD均小于1.9%,表明供试品溶液在48 h内稳定性良好。
2.4.5 加样回收试验 取已知含量的芍药甘草汤物质基准冻干粉6份,每份约0.05 g,精密称定,分别精密加入混合对照品溶液适量,按“2.3.2”项下方法制备供试品溶液,按“2.1”项下色谱条件进样测定,结果芍药内酯苷、芍药苷、芹糖甘草苷、甘草苷、异甘草苷、甘草素、苯甲酰芍药苷、甘草酸的平均加样回收率分别为100.02%、
99.26 %、100.91%、99.58%、99.29%、97.97%、93.83%、102.88%,RSD分 别 为0.40%、0.32%、0.79%、0.40%、0.67%、0.87%、0.92%、3.5%。
2.5 QAMS的建立
2.5.1 相对校正因子的计算 以芍药苷为内标物,采用多点校正法计算相对校正因子[11-12]。fi/s=fi/fs=CiAs/AiCs,公式中Ci为待测成分的质量浓度,Ai为待测成分峰面积,As为内标物对照品峰面积,Cs为内标物对照品浓度。取同一混合对照品溶液,分别进样5、10、15、20 μL,记录8种成分色谱峰面积,计算芍药内酯苷、芹糖甘草苷、甘草苷、异甘草苷、甘草素、苯甲酰芍药苷、甘草酸的fi/s分别为1.146、0.756、0.532、0.850、0.311、0.634、3.656。
2.5.2 相对校正因子耐用性考察
① 仪器和色谱柱对相对校正因子的影响:考察不同色谱柱(Dikma diamonsil C18,Thermo Syncronis C18,Agilent zorbax SB-C18,规格均为250 mm×4.6 mm,5 μm) 在Thermo U3000和Agilent 1260色谱仪上的相对校正因子,结果各成分相对校正因子的RSD<3.0%,表明仪器和色谱柱对各成分的相对校正因子无显著影响。
② 流速对相对校正因子的影响:考察不同流速(0.8、1.0、1.2 mL·min-1)下各化合物的相对校正因子,结果各成分相对校正因子的RSD<2.0%,表明流速对各成分相对校正因子无显著影响。
③ 柱温对相对校正因子的影响:考察不同柱温(25、30、35℃)下各化合物的相对校正因子,结果各成分相对校正因子的RSD<3.0%,表明柱温对各成分相对校正因子无显著影响。
2.6 待测成分色谱峰的定位
相对保留时间法是QAMS法普遍采用的色谱峰定位方法[13-15]。以相对保留时间进行色谱峰定位时,芍药内酯苷、芍药苷、芹糖甘草苷、甘草苷和异甘草苷5个待测成分的RSD小于3%,苯甲酰芍药苷和甘草酸的RSD超过了5%,表明相对保留时间对于距内参峰芍药苷较远的峰的定位效果较差。若采用两点校正法,选取芍药苷和甘草酸进行两点校正[16-17],仅有个别数据RSD大于5%,定位效果较好。
2.7 QAMS法与其他测定方法结果的比较
将15批芍药甘草汤物质基准供试品溶液注入高效液相色谱仪测定,分别采用标准曲线法、外标一点法和QAMS法计算各待测成分的含量,结果见表3。采用SPSS 21.0进行单因素方差分析,差异无统计学意义(P>0.05)。经过统计学检验,结果显示通过标准曲线法和QAMS法得到的含量之间无明显差异,说明QAMS法在芍药甘草汤物质基准含量测定研究中基本可行。
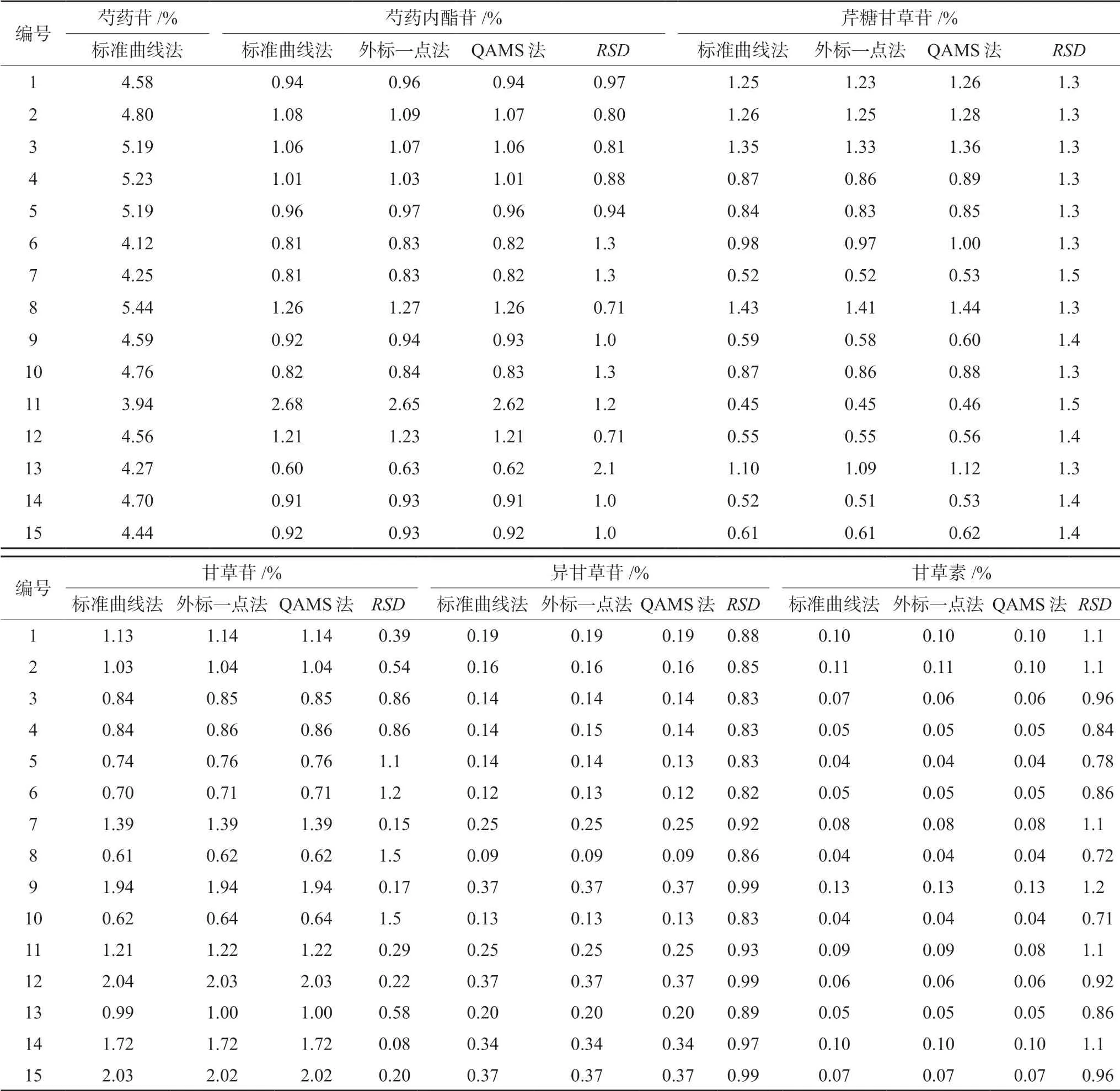
表3 8种成分不同方法含量测定结果 Tab 3 Content of 8 components by different methods
2.8 指纹图谱的建立与相似度分析
2.8.1 指纹图谱的建立 将15批芍药甘草汤物质基准的色谱数据导入“中药色谱指纹图谱相似度评价系统(2012版)”,以S1色谱图为参照图谱,中位数法谱峰匹配,确定了13个共有峰,指认出8个共有峰,分别为4号峰芍药内酯苷、5号峰芍药苷、7号峰芹糖甘草苷、8号峰甘草苷、10号峰异甘草苷、11号峰甘草素、12号峰苯甲酰芍药苷、13号峰甘草酸,见图1和图2。
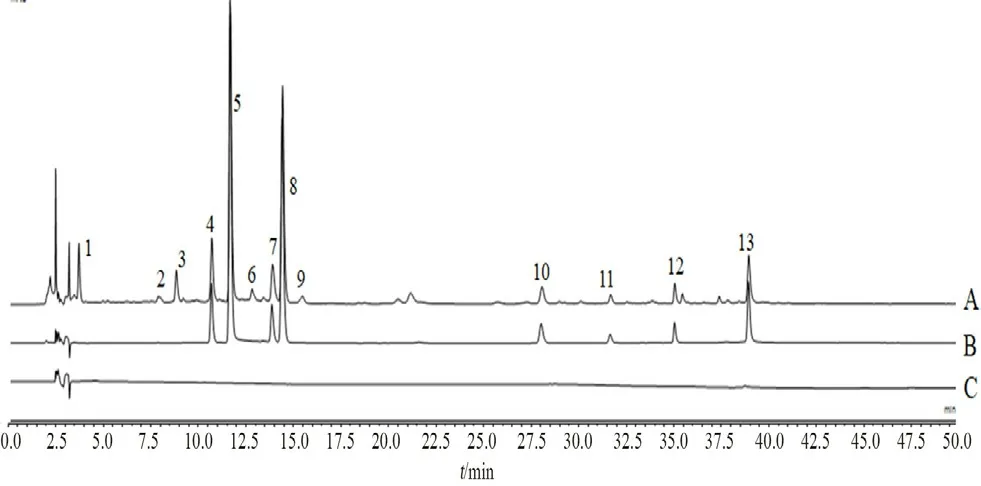
图1 样品(A)、混合对照品(B)和空白溶剂(C)HPLC图Fig 1 HPLC chromatogram of sample(A),mixed reference substances(B)and solvent blank(C)
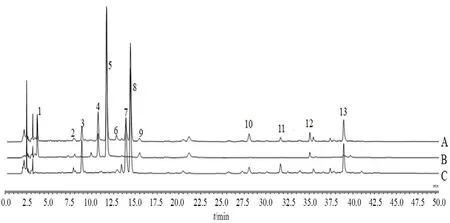
图2 样品(A)、缺甘草阴性样品(B)和缺白芍阴性样品(C)HPLC图Fig 2 HPLC chromatograms of sample(A),negative sample without licorice substances(B)and negative sample without Radix Paeoniae Alba(C)
2.8.2 芍药甘草汤指纹图谱相似度评价 对15批芍药甘草汤物质基准指纹图谱进行相似度评价,结果显示15批样品相对于对照图谱的相似度 分 别 为0.994、0.995、0.987、0.995、0.987、0.986、0.984、0.975、0.961、0.984、0.950、0.966、0.990、0.981、0.962,均大于0.96,表明15批物质基准之间差异性较小,样品叠加图见图3。
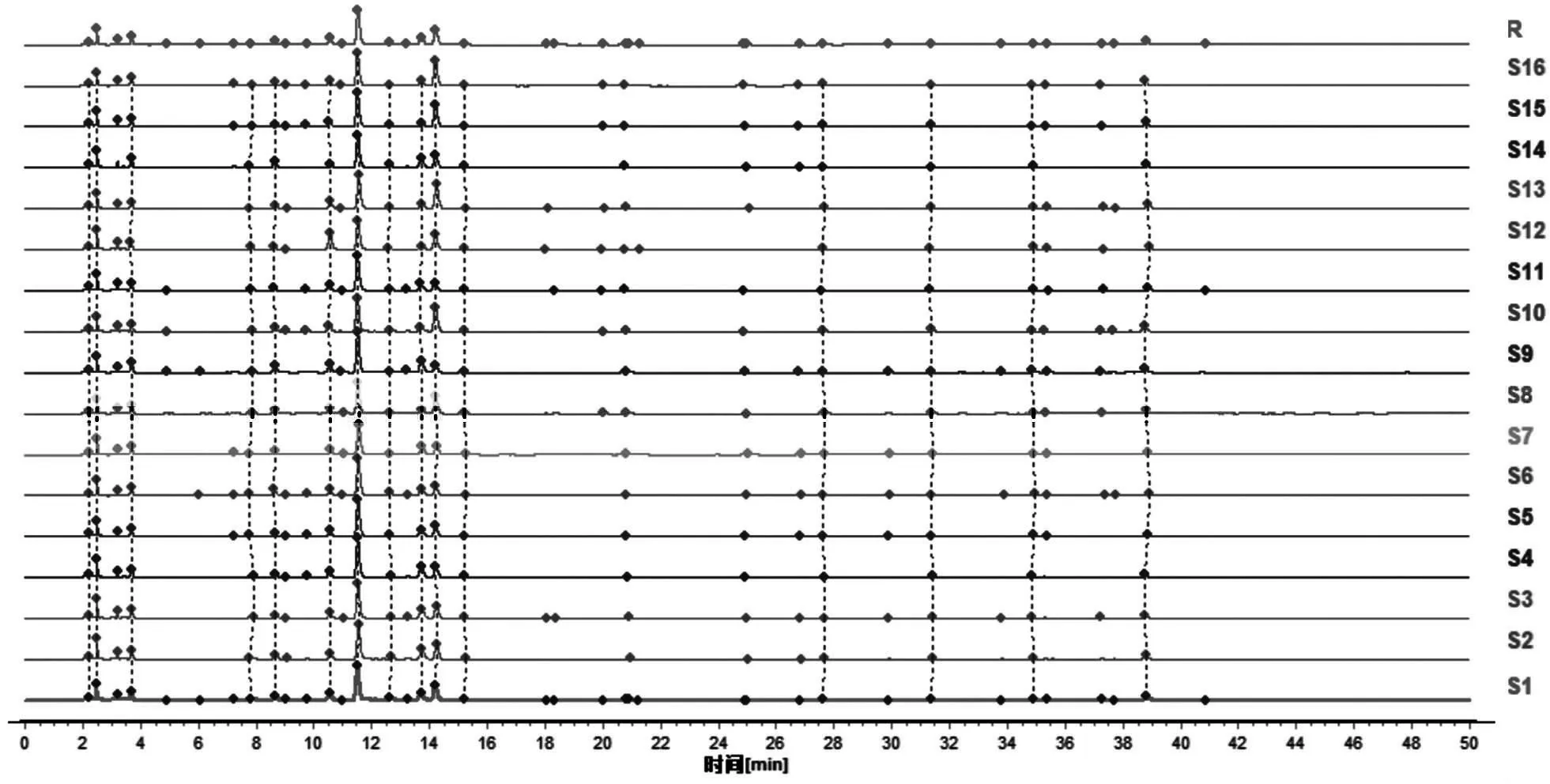
图3 15批次芍药甘草汤物质基准HPLC图及对照指纹图谱Fig 3 HPLC chromatograms and reference fingerprint chromatogram of the substance benchmarks of Shaoyao Gancao decoction

续表3
3 讨论
本研究采用多点校正法计算相对校正因子,有关文献记载采用斜率校正法,即以标准曲线斜率比值计算相对校正因子,当标准曲线线性关系较好,斜率与截距之比大于100时,斜率校正法计算得到的校正因子才具有较好的重复性和准确性[18-19]。
有文献认为,考察重现性以RSD<5%的峰进行定位[20]。但以相对保留值或保留时间差来定位时,保留时间较长的成分理论值与实测值RSD较大,并不能准确定位。本试验采用两点校正法,选用内参物芍药苷和保留时间靠后的甘草酸进行两点校正,能得到较好的定位效果。
本研究采用QAMS法,同时测定芍药甘草汤物质基准中芍药内酯苷、芍药苷、芹糖甘草苷、甘草苷、异甘草苷、甘草素、苯甲酰芍药苷和甘草素8个成分。在含量测定的基础上,建立指纹图谱,15批样品指纹图谱相似度均在0.96以上,表明不同批次物质基准的化学成分具有较好的一致性。QAMS法结合指纹图谱的方法能更全面地评价芍药甘草汤物质基准的质量,可为经典名方开发过程中的质量控制提供参考。
