靶向PSMA放射性小分子药物研究进展
2021-12-28穆博帅刘志博
穆博帅,徐 洋,刘志博,2
(1.北京大学 化学与分子工程学院,北京 100871;2.北京大学-清华大学生命科学联合中心,北京 100871)
前列腺癌(PCa)是常见的癌症之一,也是男性死亡的常见诱因。2020年,全球前列腺癌新增病例高达140万,死亡近40万[1]。现阶段,虽然原发性前列腺癌可以通过手术、化疗和放疗得到有效治疗[2],但仍然存在着较高的复发可能,特别是肿瘤扩散至淋巴结和骨骼后,其致死率将大幅提高。因此,前列腺癌的早期诊断和精准治疗具有重要意义。
临床上通常通过检测血液前列腺特异性抗原(PSA)水平和直肠指诊对前列腺癌进行诊断,但仍有10%~20%的PSA低表达患者未能得到确诊,这一问题有望通过高灵敏度的正电子发射计算机断层显像(PET)和单光子发射计算机断层显像(SPECT)技术有效解决。特别是基于前列腺特异性膜抗原(PSMA)靶标的放射性核素显像受到了广泛研究。
PSMA是一种由750个氨基酸组成的Ⅱ型跨膜蛋白质,包括胞内结构域、跨膜结构域和胞外结构域,在前列腺癌患者的前列腺上皮细胞中的表达是正常人的100~1 000倍,故被认为是前列腺癌诊疗的理想靶点[3-4]。111In标记的单克隆抗体ProstaScint®是美国FDA最早批准上市的靶向PSMA的前列腺癌放射性诊断药物,但由于较大的分子体积影响了其细胞膜通透性和内化速率[5]。相比之下,放射性核素标记的小分子药物具有更好的膜通透性,不仅在前列腺癌诊断中表现出良好的灵敏度,还在前列腺癌放疗中具有巨大潜力。
1 靶向PSMA小分子药物结构简介
早期靶向PSMA小分子药物主要基于2-PMPA等磷酸结构单元发展而来。虽然与PSMA具有良好的亲和力(<1 nmol/L)[6-7],但是较高的极性和不利的药代动力学性质限制了其在临床上的应用[8]。于是,基于巯基发展的小分子药物作为磷酸药物的替代品,显著增强了分子的膜通透性和口服生物利用率[9-12]。然而,临床上这些替代结构的药物分子在肿瘤细胞中的特异性摄取和代谢稳定性均不理想[8]。针对这些问题,直到脲基衍生的靶向PSMA药物问世才取得突破性进展。该类结构单元(如:Cys-urea-Glu和Lys-urea-Glu)对PSMA具有良好的特异选择性,基于此结构发展新型放射性药物用来诊疗前列腺癌已成为目前重要的研究领域[13](图1)。本文也将重点介绍基于脲基发展而来的靶向PSMA放射性药物。
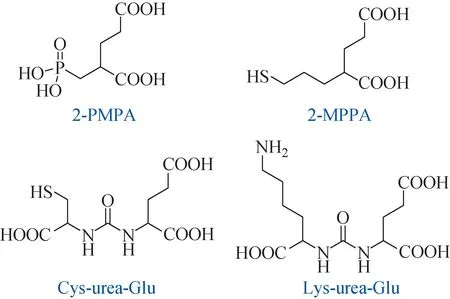
图1 靶向PSMA药物结构单元Fig.1 PSMA-targeted drug structures
2 基于脲基发展的靶向PSMA放射性药物
近些年,基于脲基结构发展而来的PSMA靶标的放射性药物受到了广泛研究。不仅实现了诊断性核素,如:11C、18F、68Ga、99mTc、123I、125I、64Cu对PSMA药物的成功标记,还发展了诸如131I、177Lu、225Ac、213Bi和212Pb等治疗性核素标记的PSMA药物(表1),下面对这两类核素标记的PSMA靶标的小分子药物分别进行介绍。

表1 基于脲基发展的靶向PSMA放射性药物Table 1 Urea-based PSMA-targeted radiopharmaceuticals
3 诊断性核素标记的靶向PSMA药物
用于诊断的放射性药物通常具有较高的检测灵敏度,仅需少量剂量即可通过PET或SPECT显像技术对患者进行无创精准的诊断[14]。早期,PSMA靶标的放射性药物的相关研究也主要集中于前列腺癌的诊断。
3.1 11C标记的靶向PSMA药物
首例基于脲基发展而来的靶向PSMA放射性药物[11C]MCG/[11C]DCMC是由Pomper等[15]在2002年报道(图2)。起初该分子主要应用于谷氨酸羧肽酶Ⅱ(GCP Ⅱ)的PET显像中。直到2005年,该药物才应用于前列腺癌的诊断中。小鼠体内实验显示,[11C]MCG在前列腺癌细胞LNCaP(PSMA阳性)中具有良好的特异性摄取,肿瘤与正常组织摄取比(T/NT)达10.8;在乳腺癌细胞(PSMA阴性)和前列腺癌细胞PC-3(PSMA阴性)中有较低的摄取[16]。虽然该药物后续也应用于临床PET诊断,但受限于11C半衰期仅为20.4 min,以及其他放射性核素的发展,11C-PSMA药物的发展变得缓慢。
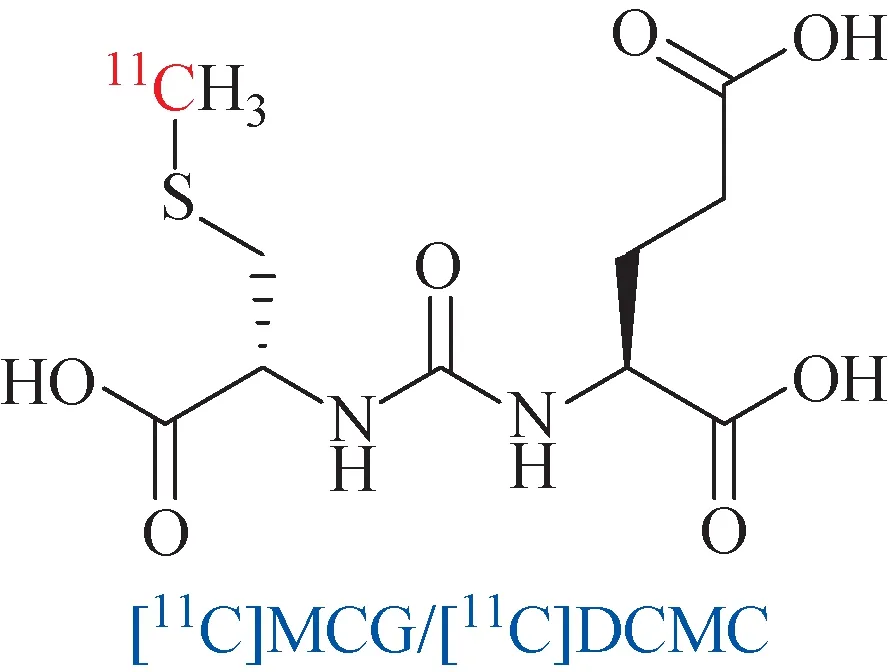
图2 11C标记的靶向PSMA药物Fig.2 11C-labeled PSMA-targeted drug
3.2 18F标记的靶向PSMA药物
相比于11C,放射性核素18F具有更长的半衰期(109.8 min),方便由加速器制备使用。18F主要通过正电子发射衰变(97%),平均能量为0.25 MeV,最大能量为0.64 MeV,在组织中湮灭距离短(约2.4 mm),可以得到高分辨率的图像,因此广泛用于临床PET显像。2008年,Mease等[7]基于[11C]DCMC的结构,合成了首例18F标记的PSMA靶标药物[18F]DCFBC(图3a)。小鼠PET显像结果显示,PC3-PIP细胞对[18F]DCFBC有较高的特异性摄取(8.16±2.55)% ID/g,在除肾脏和膀胱外的非靶组织中清除较快。但后续临床研究表明,[18F]DCFBC与血清蛋白有较强的亲和力,导致血液清除速率较慢,T/NT降低[18]。为了改善[18F]DCFBC的药代动力学性质,Chen等[19]于2011年合成了[18F]DCFPyL(图3b)。该药物不仅在阳性肿瘤中有很高的摄取(16.0±2.9)% ID/g,并且在非靶组织中能够快速清除,有望成为新的前列腺癌PET诊断药物。但在制备过程中,[18F]DCFPyL放化产率仅为(2.8±1.2)%。为了更加高效地进行18F标记,Harada等[20]使用[18F]SFB标记试剂,以30%~50%的放化产率以及大于95%的放化纯度成功地合成了四种18F标记的PSMA靶标小分子药物。其中[18F]FSU880(图3c)与PSMA的结合常数为2.23 nmol/L,且在前列腺癌细胞中的摄取可以达到(14.0±3.1)% ID/g,接近[18F]DCFPyL。
受PSMA-617(3.3节介绍)的启发,Cardinale等在其结构上引入谷氨酸残基来增加分子的亲水性,再与氟烟酸缩合合成了[18F]PSMA-1007(图3d),但放化产率仅为25%[21]。这一问题被后续发展的高效18F标记方法有效解决,放化产率可以提高到80%[22]。相比于[18F]DCFPyL,[18F]PSMA-1007具有更快的肾脏和膀胱清除速率,但却以更高的肝脏摄取作为代价,这可能是由于[18F]PSMA-1007具有较高的亲脂性所致。此外,相关临床实验还对[18F]DCFPyL和[18F]PSMA-1007进行了比较。12名患者被分成两组分别注射[18F]DCFPyL和[18F]PSMA-1007,结果显示在原位肿瘤、淋巴结转移肿瘤和骨转移肿瘤的显像效果上均有很高的灵敏度,但是[18F]DCFPyL在肾脏、膀胱和泪腺中有较高的摄取,而[18F]PSMA-1007则在肌肉、脾脏、胰腺、肝脏和胆囊等中摄取更高。因此,[18F]DCFPyL更侧重于晚期肝脏转移肿瘤诊断,而[18F]PSMA-1007在检测肾脏等泌尿系统肿瘤转移上具有更高的灵敏度[23]。
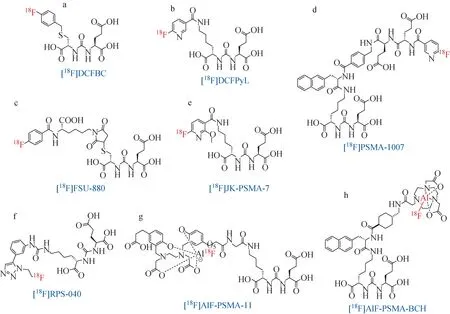
图3 18F标记的靶向PSMA药物Fig.3 18F-labeled PSMA-targeted drugs
基于[18F]DCFPyL的结构,Zlatopolskiy等[24]利用其课题组发展的18F标记的“极简方法”(minimalist approach)合成了8种[18F]JK-PSMA药物。其中,[18F]JK-PSMA-7(图3e)表现出最好的细胞活性,与LNCaP C4-2细胞的亲和力高于[18F]DCFPyL,并且在血液中的清除速率高于[68Ga]Ga-PSMA-11和[18F]PSMA-1007,因此具有更好的显像分辨率。临床研究显示[25],10名前列腺癌患者接受诊断,均未出现不良反应,检出率约为60%。并且,随着注射后时间延长,该药物表现出更理想的T/NT,为后续相关临床实验提供了依据。
2017年,Kelly等[26]还利用点击化学合成了含有三氮唑结构的药物[18F]RPS,放化产率提高为20%~40%,放化纯度大于99%。其中,[18F]RPS-040(图3f)与LNCaP细胞有更高的亲和力;小鼠PET显像实验中,[18F]RPS-040具有优秀的T/NT。为了进一步提高放射性标记效率,Boschi等[27]利用[18F]AlF标记方法以50%的放化产率和97%的放化纯度一步合成了[18F]AlF-PSMA-11(图3g)。但是[18F]AlF-PSMA-11在体内稳定性略差,脱氟副反应会影响对骨转移的检测。Liu等[28]基于该标记方法发展了[18F]AlF-PSMA-BCH(图3h),与22Rv1细胞的亲和力为(2.90±0.83)nmol/L。小鼠PET显像显示,22Rv1肿瘤对[18F]AlF-PSMA-BCH有良好的特异性摄取,能够明显区分于PC-3肿瘤;对11名前列腺癌患者的临床试验中,共检测出37处肿瘤病灶。此外,基于[18F]AlF标记方法发展的[18F]AlF-NOTA-DUPA-Pep[29],[18F]AlF-P16-093[30]等,在前列腺癌细胞显像中均具有较好效果。
3.3 68Ga标记的靶向PSMA药物
与18F相比,68Ga的优势在于通过与螯合剂作用实现标记,进而与177Lu等治疗性核素联用,实现前列腺癌的“诊疗一体化”。68Ga的半衰期为68 min,主要通过正电子发射衰变(87.94%),平均能量为0.89 MeV,最大能量为1.9 MeV。目前68Ga可以通过商业化的68Ge/68Ga发生器经过盐酸洗脱方便获得。近些年,68Ga标记的靶向PSMA小分子药物也得到了较好的发展。
2012年,Eder等[31]基于Lys-urea-Glu结构设计合成了68Ga标记的PSMA靶标药物[68Ga]Ga-PSMA-11(图4a)。该药物与PSMA有良好的亲和力,IC50为(7.5 ± 2.2)nmol/L,Ki为(12.0±2.8)nmol/L。临床实验表明,对319名前列腺癌患者的诊断检出率高达82.8%;根治性前列腺切除术后的PSA检出率为64.5%[32];特别是对于PSA低水平表达的前列腺癌患者的诊断效果也明显优于18F-Fluoromethylcholine。该药物目前已获美国FDA批准上市,主要用于前列腺癌的初期和复发性诊断。遗憾的是[68Ga]Ga-PSMA-11螯合基团并不适合与治疗性核素(90Y和177Lu等)螯合,限制了该药物在临床治疗上的研究。针对这一问题,Weineisen等[33]于2014年通过引入DOTA和DOTAGA螯合剂成功合成了DOTA-FFK(Sub-KuE)、DOTAGA-FFK(Sub-KuE),并发现螯合剂的类型对药物的生物活性有明显的影响。其中,68Ga标记的DOTAGA-FFK(Sub-KuE)以及其非对映异构体DOTAGA-ffk(Sub-KuE)均能够表现出比DOTA-FFK(Sub-KuE)更优的亲和力、体内代谢稳定性和更快的细胞内化速率(图4c)。除此之外,[177Lu]Lu-DOTAGA结构的药物也同样表现出良好的生物活性,这为诊疗结合的放射性药物开发提供了有利依据。在此基础上,该课题组通过对结构的进一步优化,合成了[68Ga]Ga-PSMA-I&T(图4b)[31]。该药物与PSMA的亲和力同样可达到纳摩尔水平,且体内肿瘤靶向性良好,代谢速率快。临床PET诊断实验中,[68Ga]Ga-PSMA-I&T能够以高肿瘤背景比跟踪骨、淋巴结以及肝脏肿瘤转移,适用于转移性及去势性前列腺癌的诊断。
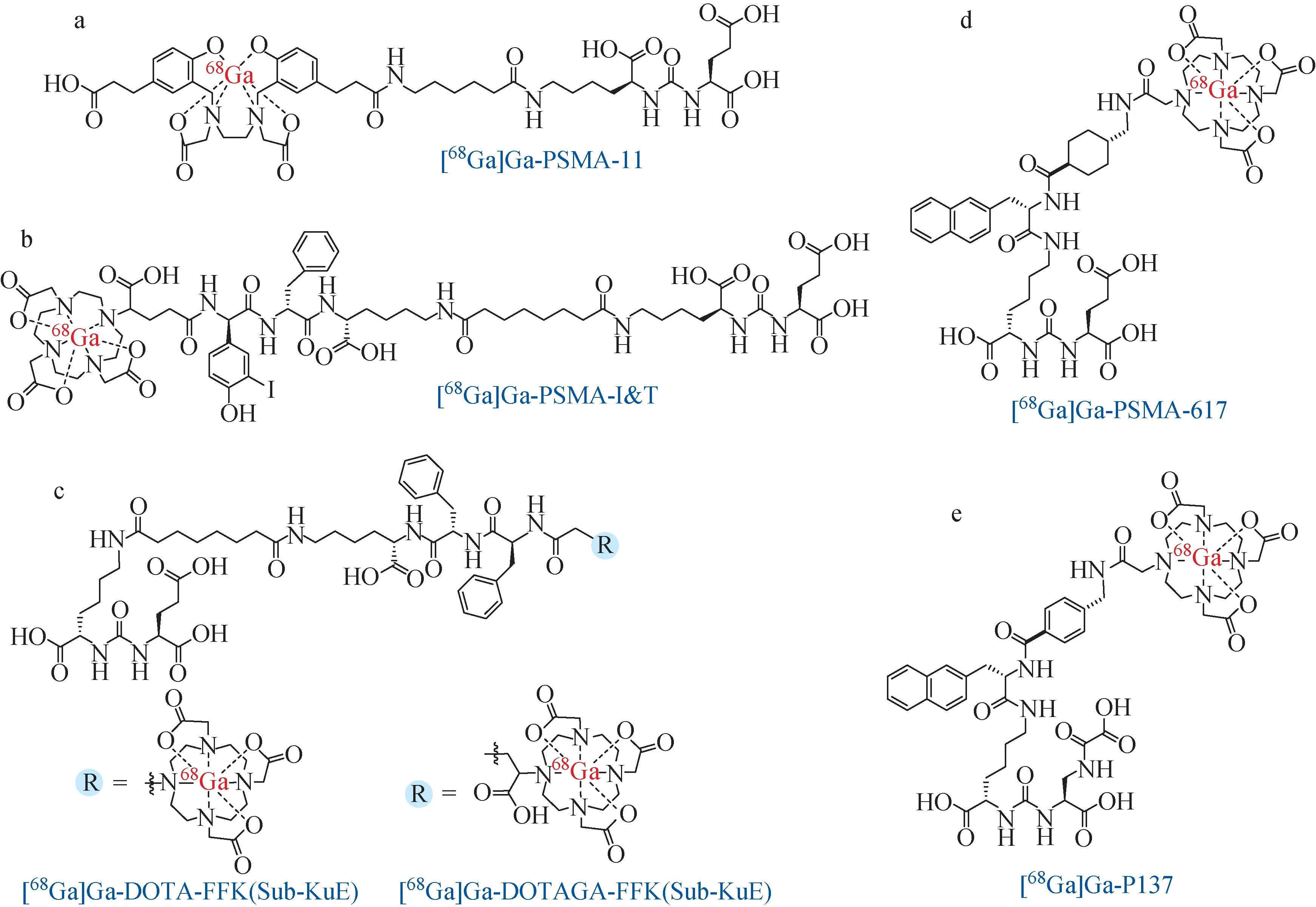
图4 68Ga标记的靶向PSMA药物Fig.4 68Ga-labeled PSMA-targeted drugs
2015年,Benešová等[34]报道的含有DOTA螯合基团的[68Ga]Ga-PSMA-617(图4d)同样具有理想的T/NT和良好的阳性肿瘤特异性摄取。相应的[177Lu]Lu-PSMA-617在前列腺癌临床治疗上也具有突出效果。值得一提的是,[68Ga]Ga-PSMA-617的标记过程需要在加热和酸性条件下才能进行,不利于药物分子的官能团兼容性。为了更加高效地进行标记实验,Young等[35]设计合成了THP-PSMA,在室温、中性条件下即可高效实现68Ga标记。相关细胞和体内实验表明,[68Ga]Ga-THP-PSMA对肿瘤组织具有良好的特异选择性,细胞内化、体内代谢速率与生物分布均达到了与[68Ga]Ga-PSMA-11相似的水平。2021年,Duan等[36]基于ODAP-urea的结构成功合成了12种小分子药物,其中[68Ga]Ga-P137(图4e)凭借优秀的体内外稳定性、高PSMA亲和力和肿瘤细胞22Rv1高特异性摄取被用于临床研究。结果表明,相比[68Ga]Ga-PSMA-617,[68Ga]Ga-P137有更好的药代动力学性质,进一步证实了ODAP-urea药效团结构的可行性。
3.4 99mTc标记的靶向PSMA药物
99mTc是应用最为广泛的医用放射性核素,可通过99Mo/99mTc发生器方便获得,在SPECT显像诊断中具有重要地位。其半衰期为6 h,衰变时会释放低能量γ射线(140 keV),对生物体的辐射损伤小并且具有良好的显像分辨率。由于锝元素的价态丰富,因此可以与多种结构的螯合剂作用形成不同立体构型的配合物,有利于相关放射性药物的设计与开发。
首例99mTc标记的靶向PSMA小分子药物由Mishra等于2007年报道[37]。他们基于磷酸酯结构设计合成了三种含有金刚烷基的99mTc标记的PSMA药物。其中三聚结构的[99mTc]Tc-MAS3-IVa(图5a)与PSMA的亲和力最高,半数抑制浓度可达3 nmol/L,并且在血清中具有很高的稳定性。随后,Lu等[38]基于Lys-urea-Glu结构单元设计合成了一系列含有吡啶和咪唑螯合基团的靶向PSMA小分子药物,并成功实现99mTc的标记。其中,含有四羧基螯合基团(TIM)的[99mTc]Tc-MIP-1404和[99mTc]Tc-MIP-1428(图5b)不仅在生物体外和体内均表现出对PSMA良好的特异选择性和亲和力,还能够表现出更快的肾脏及非靶组织的代谢速率[39]。相关临床实验完成了对471名无前列腺癌临床症状患者的诊断,结果显示[99mTc]Tc-MIP-1404的特异性能够达到71%~75%,但是灵敏度仍有待于提高[40-43]。
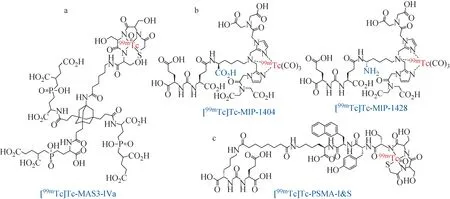
图5 99mTc标记的靶向PSMA药物Fig.5 99mTc-labeled PSMA-targeted drugs
2013年,Bay和Banergee等[44]利用不同99mTc标记方法,系统研究了螯合剂对PSMA靶标药物生物活性的影响。通过考察不同价态[99mTc(CO)3]+、[99mTcO]3+和[99mTcNHNR]2+标记的12种小分子药物,发现[99mTc(CO)3]+标记的药物对肿瘤有着最高的摄取量,并且在包括肾脏等非靶组织内的滞留时间最短,具有进入相关临床实验的潜力。
随着[111In]In-PSMA-I&T在放射性显像导向手术(radioguided surgery,RGS)中的应用[45],2017年Wirtz等[46]报道了具有相似结构的PSMA放射性探针[99mTc]Tc-PSMA-I&S(图5c)。该99mTc探针不仅能够方便地利用发生器制备降低使用成本,还具有与[111In]In-PSMA-I&T相同水平的LNCaP细胞内化效率,且亲水性提高了一个数量级。在临床SPECT显像中,[99mTc]Tc-PSMA-I&S的全身清除速率较慢,导致5 h之后才获得优秀的T/NT。基于以上人体实验数据,放射性显像导向手术在患者注射[99mTc]Tc-PSMA-I&S后16 h进行;注射12 h后的SPECT显像能够清晰的发现肿瘤向淋巴结转移,有效实现了对病灶部位的精准识别和切除。
3.5 放射性碘同位素标记的靶向PSMA药物
放射性碘同位素包括123I、124I、125I和131I,它们的性质和制备方法存在差异,在核医学中有着不同的应用范围。其中,123I和124I分别应用于SPECT和PET显像;125I可用于近距离放射性治疗;而131I则既可用于SPECT显像又可以应用于放射治疗。
放射性碘同位素标记的靶向PSMA药物由Pomper课题组在2005年首次报道[16]。他们基于Lys-urea-Glu结构单元合成了[125I]DCIT(图6a),尽管与PSMA有着良好的亲和力(1.5 nmol/L),但病灶部位的清除速率明显快于阳性对照分子[11C]DCMC,不利于肿瘤诊断。后续研究中,该课题组又发展了一种[125I]DCIBzl药物(图6b),与PSMA的亲和力达到10 pmol/L,这也成为PSMA靶标药物开发过程中理想的阳性对照[47]。
2009年,Maresa等[48]基于Lys-urea-Glu结构单元合成了一系列小分子药物,通过细胞实验的筛选发现,碘取代的MIP-1072和MIP-1095的半数抑制浓度均可以达到纳摩尔水平,遂对其进行放射性核素标记,成功获得了相应的[123I]MIP-1072和[131I]MIP-1095(图6c)。随后的临床SPECT显像中,[123I]MIP-1072具有高达20的肿瘤背景比。并且,[123I]MIP-1072在除膀胱外的非靶组织摄取量均低于[131I]MIP-1095,而膀胱的摄取量却是[131I]MIP-1095的4倍,这也是药物快速通过尿液代谢的有利信号。果然血液清除实验显示,[123I]MIP-1072具有更快的血液清除速率[49]。由于131I可用于放射治疗的核素,[131I]MIP-1095也成为了首个PSMA靶标的用于临床放疗实验的药物。28名去势抵抗性前列腺癌患者接受了[131I]MIP-1095的注射(平均放射性活度:4.8 GBq),其中摄取剂量最高的非靶器官分别为唾液腺(3.8 mSv/MBq)、肝脏(1.7 mSv/MBq)和肾脏(1.4 mSv/MBq)。首轮放疗后,60.6%的患者PSA水平降低了50%以上;84.6%的骨痛患者有了完全或明显的缓解;仅有25%的患者出现口干症状;患者肾脏均未见明显不适[50]。遗憾的是,虽然首轮治疗取得了较好的效果和较小的药物副作用,但是随后的治疗周期并未取得进一步的疗效[51]。
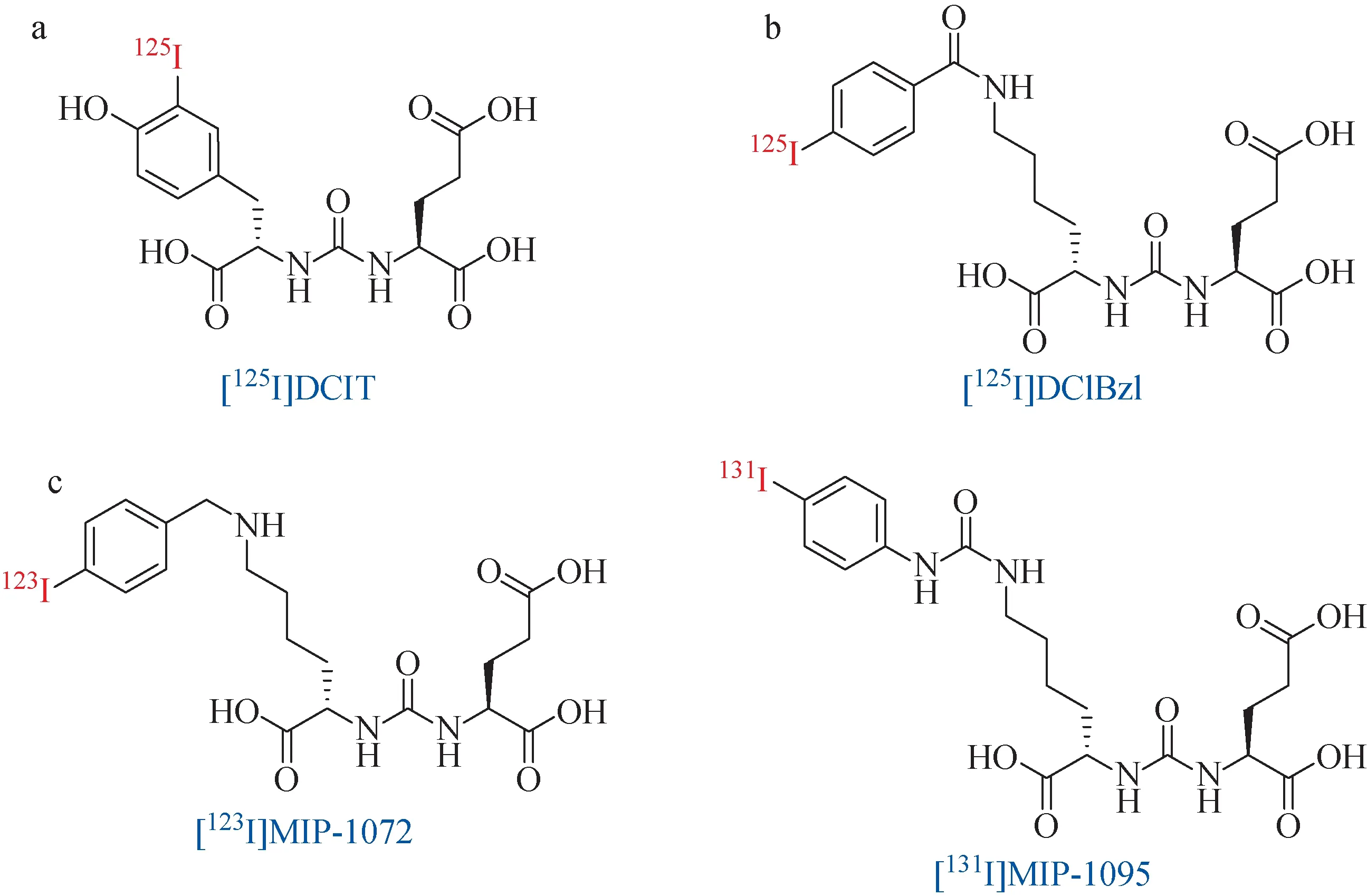
图6 放射性碘同位素标记的靶向PSMA药物Fig.6 Radioiodine labeled PSMA-targeted drugs
3.6 64Cu标记的靶向PSMA药物
放射性核素64Cu主要通过反应堆或粒子加速器两种途径制备,半衰期为12.7 h,适合于放射性药物中心批量化制备及发运,其衰变性质(β+17.4%,Emax=0.655 MeV,β-39.6%,Emax=0.573 MeV以及电子俘获)使其不仅可应用于PET显像,还在放疗中具有一定的应用前景。
2016年,Grubmüller等[52]报道了首例[64Cu]Cu-PSMA-617在PET/CT临床诊断中的研究。结果显示该药物在前列腺原发肿瘤或复发性肿瘤均有较高的摄取,29名患者在给药1 h后就能够以良好的T/NT检测出肿瘤病变,其中有23名被探测到至少1个病灶,即使在PSA低水平表达患者中也具有很高的检出率。并且,接受治疗的患者均未出现不良反应。但是次年的一项工作[53]指出该药物在体内稳定性较差,有较高的肝脏摄取量和较慢的肾脏清除速率,该现象在之前合成的64Cu-DOTA型药物中也有出现[54]。分析认为64Cu不再与DOTA配位,转而与肝脏中的超氧化物歧化酶(SOD)结合[55],从而造成肝脏放射活性的积累。因此,为提高肿瘤肝脏比,需要使用其他螯合剂取代DOTA,如CB-TE2A[56],NODAGA[57],NODIA-Me[58],AAZTA[59]等。
2014年,Banerjee等[56]利用五种不同的大环螯合剂NOTA、PCTA、oxo-DO3A、CB-TE2A、DOTA分别合成了相应64Cu标记的小分子药物。小鼠PET显像表明,CB-TE2A衍生的64Cu-PSMA型药物在PC3-PIP细胞中摄取最高,且在非靶向组织中清除速率最快。这一结果与CB-TE2A和Cu的稳定螯合作用密不可分。2015年,Gourni等[57]合成了[64Cu]Cu-CC34(图7b)。体内生物学分布显示,[64Cu]Cu-CC34在非靶组织中积聚少、清除快、肿瘤背景比高,表明NODAGA能与64Cu稳定配位,有较高的体内稳定性。Ghosh等[60]也通过体内外实验证明螯合剂NODAGA相比DOTA更有应用前景。

图7 64Cu标记的靶向PSMA药物Fig.7 64Cu-labeled PSMA-targeted drugs
2020年,Santos等[61]基于PSMA-617结构发展了[64Cu]Cu-CA003,并与[64Cu]Cu-PSMA-617进行了比较。小鼠体内实验表明,[64Cu]Cu-CA003有较低的肝脏摄取、较高的体内稳定性、较快的肾脏清除和较好的显像对比,有望取代[64Cu]Cu-PSMA-617成为新的临床候选药物。此外,Liu等在[18F]AlF-PSMA-BCH的基础上设计的药物[64Cu]Cu-PSMA-BCH也有较高的体内稳定性,但仍需更多的临床前研究和临床研究证明其有效性[62]。
为更好地探测小转移瘤,Müller课题组设计合成了一系列白蛋白结合剂修饰的PSMA靶标药物[64Cu]Cu-PSMA-ALB,该类药物特别是[64Cu]Cu-PSMA-ALB-89(图7c)有较长的血液循环时间,能够提高小转移瘤部位的放射活性[63]。受到上述工作启发,Babich课题组[64-66]在MIP-1095的基础上合成了RPS系列小分子药物。其中,[64Cu]Cu-RPS-085[67]在静脉注射4 h后,表现出比[64Cu]Cu-PSMA-ALB-89更高的T/NT,有很好的应用前景。
4 治疗性核素标记的靶向PSMA药物
目前,用于PSMA靶标的治疗性核素主要包括131I、177Lu、225Ac、213Bi和212Pb等。随着68Ga-PSMA型药物在临床诊断中的应用,一些效果较好的药物分子也被用于肿瘤的放疗。
4.1 177Lu标记的靶向PSMA药物
前文提到的[131I]MIP-1095是基于脲基发展的最早用来放射性治疗前列腺癌的小分子药物。但是131I所产生的γ辐射较强,对肿瘤细胞以外的正常细胞也有杀伤作用,导致其毒副作用相对较强。而177Lu作为一种β放射性核素,其γ辐射较弱、射程较短,因此对正常细胞的杀伤力较弱,并且其半衰期长达6.7 d,是较为理想的用于放射性治疗的核素[68]。目前,用于临床研究的靶向PSMA药物[177Lu]Lu-PSMA-I&T(图8a)和[177Lu]Lu-PSMA-617(图8b),在临床实验中均可大幅降低前列腺癌患者的PSA水平,有很好的抗肿瘤活性和安全性[69]。
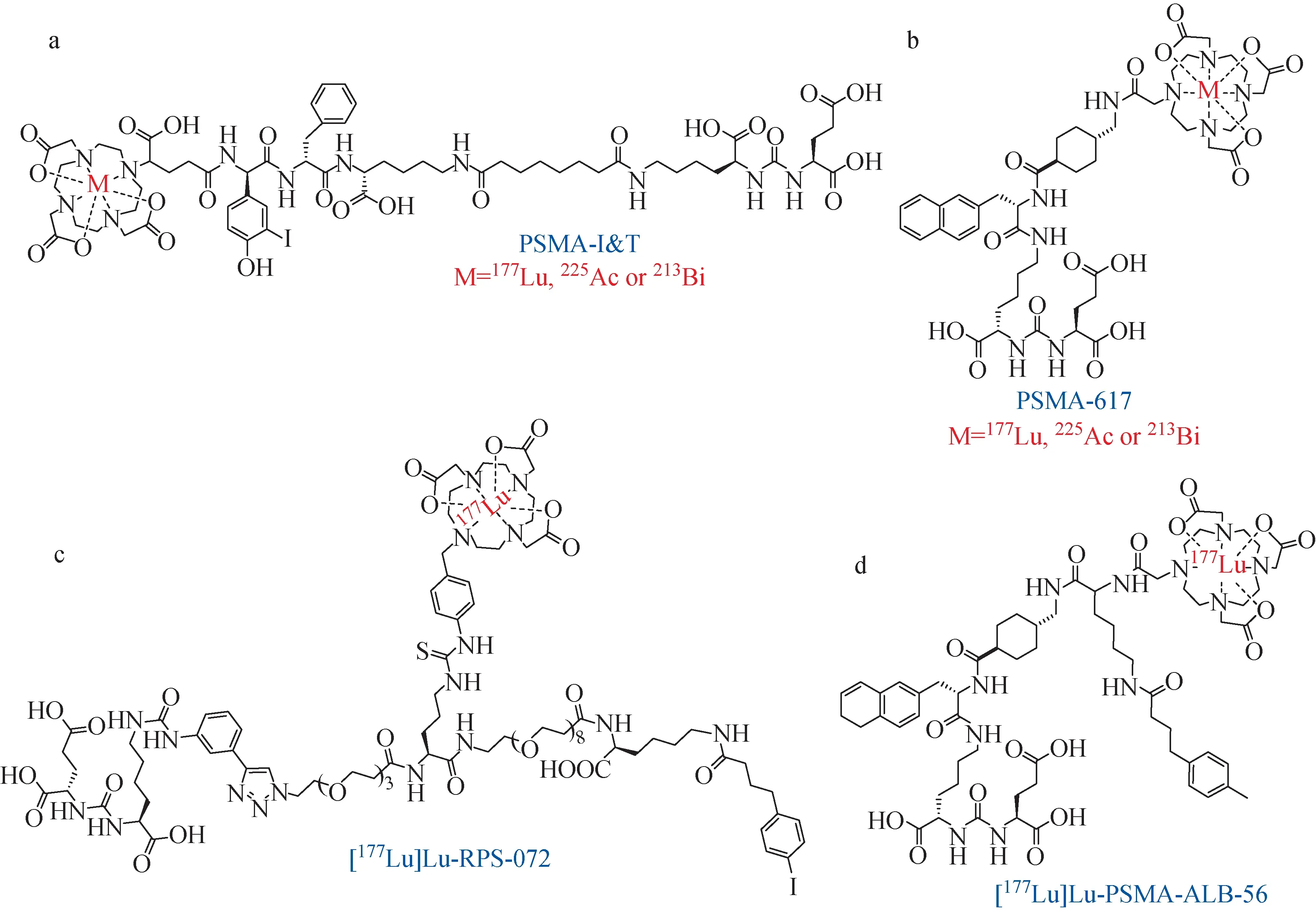
图8 177Lu、225Ac和213Bi标记的靶向PSMA药物Fig.8 177Lu-/225Ac-/213Bi-labeled PSMA-targeted drugs
2015年,Eder等[34]与Weineisen等[32]分别合成了[177Lu]Lu-PSMA-617和[177Lu]Lu-PSMA-I&T药物。最近,一项临床前研究对这两种药物进行了系统的比较[70]。体外实验结果显示,三种阳性肿瘤细胞(PC295,PC82,PC310)对[177Lu]Lu-PSMA-617和[177Lu]Lu-PSMA-I&T都有较高的摄取,而阴性肿瘤细胞(PC324)基本无摄取。小鼠体内生物学分布结果显示,肿瘤部位对两种药物都有较高摄取,并且随着时间的推移,T/NT不断提高。但是,[177Lu]Lu-PSMA-I&T在肾脏中的积聚远高于[177Lu]Lu-PSMA-617。随着诺华(Novartis)加入对[177Lu]Lu-PSMA-617的临床研究,该药物分子依然成为了当下的明星分子。由于[177Lu]Lu-PSMA-617在治疗转移性去势抵抗性前列腺癌中有较理想的疗效,因此被美国FDA授予突破性药物资格(BTD)。近期,诺华公布的临床III期结果显示,[177Lu]Lu-PSMA-617联合标准护理SOC显著改善了前列腺癌患者总生存期和放射学无进展生存期[71]。
此外,Babich课题组发展的RPS系列药物[64-66]和Müller课题组发展ALB系列药物[72-73],也在临床前研究中表现出很好的性质,如[177Lu]Lu-RPS-072(图8c)和[177Lu]Lu-PSMA-ALB-56(图8d)均有较高的肿瘤摄取和较快的背景清除速率,有良好的应用前景,需要临床研究的进一步验证。
4.2 225Ac、213Bi和212Pb标记的靶向PSMA药物
尽管177Lu在肿瘤放疗中有很好的应用前景,但临床研究表明大约30%的患者对177Lu-PSMA型药物没有响应,同时,β辐射可能会带来血液学毒性[74]。不同于β粒子,α粒子传能线密度(LET)高、射程短(50~90 μm),能在短距离内释放出大量能量,可有效导致双链DNA断裂、染色体畸变和细胞死亡,对癌细胞具有更高的杀伤力。尽管如此,α放射性核素标记的靶向PSMA药物仅处在起步阶段。下面将介绍基于225Ac、213Bi和212Pb等α放射性核素发展的靶向PSMA放射性药物。
225Ac或213Bi标记的PSMA-617或PSMA-I&T是目前具有临床代表性的药物。关于[225Ac]Ac-PSMA-617[75-76]和[213Bi]Bi-PSMA-617[77]的临床研究显示,二者均在前列腺癌的治疗上具有理想的效果,使患者体内PSA水平明显下降。但也有数据表明[213Bi]Bi-PSMA-617在治疗过程中出现脱靶的情况,有对非靶组织杀伤的潜在弊端[78]。
最近,Zacherl等[79]报道了[225Ac]Ac-PSMA-I&T的第一例临床研究,超过70%的患者在接受治疗后,体内PSA水平出现不同程度的下降,其中部分患者先前对[177Lu]Lu-PSMA-I&T治疗无响应。对[213Bi]Bi-PSMA-I&T的临床前研究由Nonnekens在2017年报道[80],[213Bi]Bi-PSMA-I&T在体内外实验中均可诱导双链DNA的断裂,对肿瘤细胞造成更大的杀伤。但该药物也具有肾脏摄取较高的通病,可能带来的肾毒性将成为后续临床研究需要面临的问题。除此之外,可与白蛋白结合的[225Ac]Ac-RPS-074[81]、225Ac/213Bi标记的PSMA-DA1[82]和PSMA-L1[83]也相继被报道,但相关的临床研究仍有待进行。
相比于225Ac(半衰期9.9 d)和213Bi(半衰期46 min),212Pb的半衰期较为适中(10.6 h),可以提供理想的内照射治疗周期。2019年,Dos等[84]研究了[212Pb]Pb-CA012(图9)在前列腺肿瘤治疗上的效果。患者单个治疗周期的最大耐受剂量可以达到150 MBq,并且在骨肿瘤上的特异性摄取高于[213Bi]Bi-PSMA-617,但不及[225Ac]Ac-PSMA-617,有待改善。
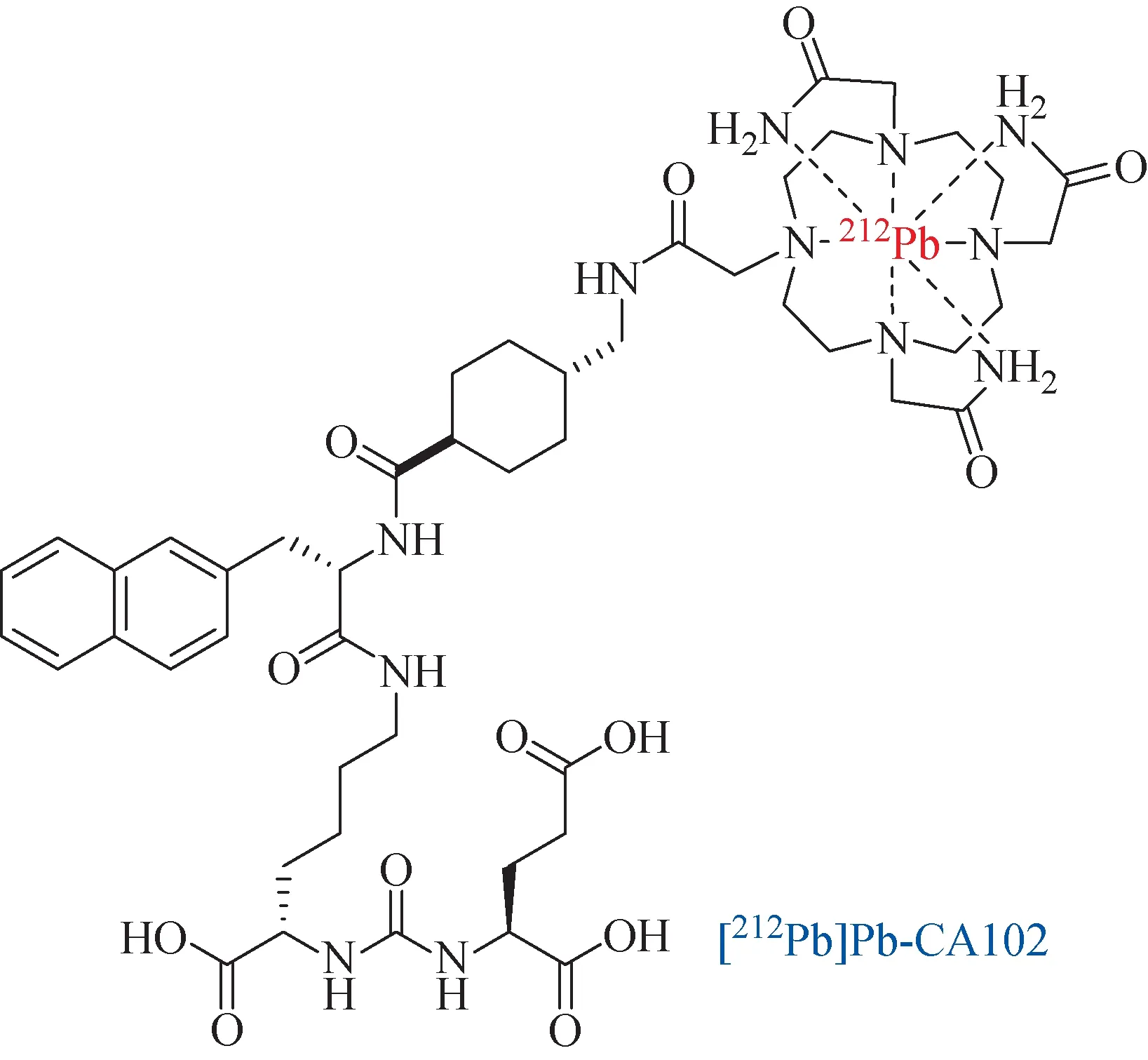
图9 212Pb标记的靶向PSMA药物Fig.9 212Pb-labeled PSMA-targeted drugs
5 总结与展望
PSMA能够在前列腺癌患者的前列腺上皮细胞中高特异性表达,是现阶段前列腺癌放射性诊断和治疗的理想靶点。基于该靶点发展的靶向PSMA放射性药物受到了广泛研究,在前列腺癌诊疗中具有广阔的应用前景。其核心结构单元主要分为磷酸衍生的2-PMPA、巯基衍生的2-MPPA以及脲基衍生的Cys-urea-Glu和Lys-urea-Glu。但基于磷酸和巯基结构发展的放射性药物由于药代动力学性质上的不足,尚未在临床研究上取得显著进展。相比之下,基于脲基发展的靶向PSMA放射性药物得到了更好的发展。例如诊断型放射性药物[68Ga]Ga-PSMA-617、[68Ga]Ga-PSMA-I&T、[68Ga]Ga-PSMA-11、[18F]DCFBC、[18F]DCFPyL和[99mTc]Tc-MIP-1404等均实现了在临床PET或SPECT医学显像中的应用。而用于治疗的放射性药物则需要更高的剂量来实现对病灶部位的有效治疗,同时还需避免过高的辐射剂量对非靶组织带来的损伤,这对药物在体内的代谢和生物分布有着更高的要求。随着含DOTA螯合剂“诊疗一体化”药物分子的发展,[177Lu]Lu-PSMA-617、[177Lu]Lu-PSMA-I&T等β放射性核素标记的药物也相继进入了临床研究。近期诺华(Novartis)公司在[177Lu]Lu-PSMA-617三期临床研究中取得了新的进展,受到了同行的广泛关注。尽管如此,二期临床研究结果显示该药物仍存在一定的唾液腺和肾毒性,部分患者出现了1级或2级口干副作用以及1级或2级肾功能紊乱副作用[85]。因此,发展新的药物结构和探索新的给药方法[86]来降低药物对非靶组织的毒性是PSMA靶标药物研发中需要改善与解决的问题。此外,α放射性核素标记的PSMA靶标药物可通过释放α射线使双链DNA断裂从而对癌细胞进行有效杀伤,特别是212Pb标记的药物,其半衰期为10.6 h,不仅可以提供有效的放射治疗时长,还可以避免半衰期过长为后处理带来的不便,是一种具有研究前景的治疗用放射性核素[87]。此外,224Ra也是一种具有良好衰变性质的α放射性核素,但目前仍需发展新型匹配的螯合剂以提高其标记药物的体内稳定性。所以,新型治疗性核素的开发以及新型螯合剂[88]的筛选是靶向PSMA放射性药物研发的重要环节,也是推动整个放射性药物研究领域不断进步的重要动力。
