南海近海浮游细菌群落结构和多样性研究
2021-12-22孙涛,付婧,周进
孙 涛,付 婧,周 进
(1.上海海洋大学水产与生命学院,上海 201306;2.中国水产科学研究院东海水产研究所,上海 200090)
南海是世界第三、太平洋西部最大的边缘海,最大深度可超过5000 m,具有复杂的物理化学环境。黑潮、季风、上升流和涡旋等物理力量的干扰较为显著地影响南海水体物理化学性质,造就其区域高度异质化的生境空间[1-3]。细菌是海洋微生物中的优势类群,广泛参与海洋生源要素的生物地球化学循环,已有研究报道,南海水域微生物多样性水平较高[4-6]。
针对南海水域细菌的物种多样性和群落结构已有较好研究基础。然而,现阶段海洋群落生态学研究多依赖于数量较为有限的现场采集,此种基于有限数量样本的群落结构描述与实际数据之间尚存差距。南海区域辽阔,海况通常较为恶劣,海洋研究样品采集成本较高。已有南海细菌群落结构和多样性研究中样品采集站位数量通常较少,站位数多为十余个至数十个[4-7],研究覆盖的区域范围较为有限,因此以大范围采样为基础的南海细菌群落研究具有重要意义。南海水体总体处于寡营养状态,珠江河口和陆域径流向海域输入的营养物质,易在海域内形成沿岸至深海渐变的细菌生境,此种渐变生境可能在较小的空间范围内具有较为明显的理化环境条件梯度[9]。揭示此种梯度化生境条件下的细菌群落结构可为理解南海环境异质性提供生物学证据。
已有针对南海细菌群落的研究多探讨表层海水中细菌群落的空间分布特点以及细菌在水体环境中的垂直分布[6]。研究发现,南海海水中细菌类群受到理化环境因素的影响,上层海水中细菌丰富度和多样性随深度增加而增加,垂直分布较为显著[10-11]。也有研究发现,南海表层细菌群落具有生态类型(沿岸和海洋区)和中尺度物理过程的两级空间分布模式[6]。以往针对南海细菌的空间分布和垂直分布特征研究,多数研究调查站位较少,采样范围较为有限[4-7],采样范围未覆盖南海东北部、琼东和北部湾等南海近岸典型水域等,或者是研究一些特定的生境,如上升流、冷泉等[12-13]。伴随分子生物学技术方法的发展,细菌的多样性及其分布格局有望得以深入研究。
本研究在南海海域实现了较大规模的海水细菌样品采集,采集范围覆盖南海东北部、琼东和北部湾等南海近岸典型水域。研究旨在描述南海近海浮游细菌多样性、群落结构及其空间分布特征,基于较大样品采集信息的分析研究有望更为科学、客观地揭示南海细菌分布格局,以期为南海微生物资源开发利用奠定理论基础。
1 材料和方法
1.1 样品采集
本研究在南海水域设置50个采样站位(图1),采样区域主要包括南海东北部、琼东和北部湾共3个区域。利用船载玫瑰花式采水器采集表层(0~5 m)和底层海水(部分站位)样品各2 L,样品通过负压过滤方法抽滤至0.22μm聚碳酸酯膜(Merck-Millipore,Iceland),滤膜样品立即转移至无菌冻存管,置于液氮中保存。样品采集搭载国家自然科学基金委和广东海洋大学组织的开放航次,样品采集时间为2018年6—8月。
1.2 基因组DNA提取与16Sr RNA高通量测序
在无菌条件下将滤膜样品剪碎,按照DNA提取试剂盒(FastDNA®Spin Kit for Soil,MP Biomedicals)规定步骤严格进行基因组DNA抽提。利用NanoDrop 2000分光光度计2000c(Thermo Fisher Scientific Inc.,Wilmington,DE,USA)检测DNA提取物的浓度(ng·μL-1),利用1%琼脂糖凝胶电泳检测DNA抽提质量。
利用16SrRNA基因V3-V4高变区序列信息鉴定微生物物种。选取10 ng高浓度和高质量DNA为模板,使用带有barcode的特异正向引物338F(5′-ACTCCTACGGGAGGCAGCAG-3′)和 反向引物806R(5′-GGACTACHVGGGTWTCTAAT-3′)扩增V3-V4 rRNA靶区域,共进行3次扩增[14]。PCR扩增反应条件为:95℃预变性3 min,循环35次(95℃变性30 s,50℃退火30 s,72℃延伸45 s);最后72℃延伸10 min,10℃保存。扩增体系(20μL):5×FastPfuBuffer 4μL、dNTPs(2.5 mmol·L-1)2μL、正向引物Forward Primer(5μmol·L-1)0.8μL、反向引物Reverse Primer(5μmol·L-1)0.8μL、FastPfuPolymerase 0.4μL、BSA 0.2μL、Template DNA 10 ng、补ddH2O至20μL。PCR产物经2%琼脂糖凝胶电泳检测,用QuantiFluorTM-ST蓝色荧光定量系统(Promega Corporation,Madison,WI,USA)进行定量检测。按照每个样本的测序量要求,进行相应比例的混合,构建Miseq文库。高通量测序由美吉生物医药科技有限公司(上海)使用Miseq PE300(Illumina Inc,San Diego,CA,USA)测序平台完成,测序策略为2×300 bp。
本研究所使用的原始序列已上传至NCBI的SRA数据库,登录号为SRX11623561-SRX11623624。
1.3 数据处理和生物信息学分析
根据overlap关系对Miseq测序得到的PE reads进行拼接,同时对序列质量进行质控拼接和过滤,去除引物序列和含barcode序列,优化测序数据。利 用Usearch[15](version 7.1 http://drive5.com/uparse/)软 件 平 台(UPARSE[16]:highly accurate OTU sequences from microbial amplicon reads)对非重复序列(不含单序列)进行聚类,根据97%的序列相似度归类产生操作分类单元(operational taxonomic unit,OTU)。聚类过程中去除嵌合体。
采用RDP classifier贝叶斯算法[17](置信度阈值为0.8)对OTU代表序列进行比对注释,对照Silva数据库[18](silva138/16s_bacteria,http://www.arb-silva.de)匹配各OTU的分类信息。匹配至叶绿体(chloroplast)、线粒体(mitochondria)、古菌(Archaea)和未归类的OTU,以及相对丰度小于序列总数0.01%的OTU未被包括在后续群落结构分析之内。
1.4 细菌群落多样性分析
本文基于α-和β-多样性两个角度描述研究区域细菌群落的结构特征和多样性水平。α-多样性分析时计算如下参数:OTU数量(OTUs)、覆盖度(Good's coverage,G)、多样性指数(Shannon-Wiener,SW)、单纯度指数(Simpson)和丰富度指数(Chao 1,Ace)。使用QIIME[19](1.9.1,http://qiime.org/install/index.html)软件运行core_diversity_analyses.py脚本文件计算α-多样性指数。数据类型不符合正态分布和方差不齐,选用Wilcoxon秩和检验(Wilcoxon rank-sum test)判别群落多样性和丰富度指数在各采样区域之间是否具有显著差异。
基于Bray-curtis非相似性系数,利用UPGMA聚类方法分析不同样品之间16SrRNA基因序列的相似性,并利用非度量多维尺度分析方法(nonmetric multidimensional scaling,NMDS)揭示不同样品之间的差异性,使用ANOSIM检验NMDS组间差异显著性。根据采集样品的细菌组成以及不同类群相对丰度绘制热图,研究水体细菌组成的空间差异。聚类和非度量多维尺度分析以及热图绘制利用R软件完成。
利用LEfSe工具采用线性判别分析方法[20-21](linear discriminant analysis,LDA)估算细菌组成对样品空间差异的影响效果,鉴定水体样品中的特异性细菌类群[13-14]。LDA值表征特定细菌类群相对丰度在不同样品间的差异程度,具有较高LDA值的细菌类群可视为群落的生物标记物。本研究以LDA>3作为生物标记物的筛选标准。
2 结果与分析
2.1 细菌群落α-多样性
本研究共采集64个水体样品。序列筛选后共获取3041767条高质量16S rRNA基因序列(97%相似水平)和2891个优化OTU。单样本序列数变化范围为34457~68506。单样本OTU数量变化范围为241~1245,11号采样站位表层水体样品中OTU数量最低,33号站位底层样品中OTU数量最高。如图2所示,曲线逐渐趋于平坦,表明本研究测序量较为合理,测序深度足以反映采样区域细菌的多样性。
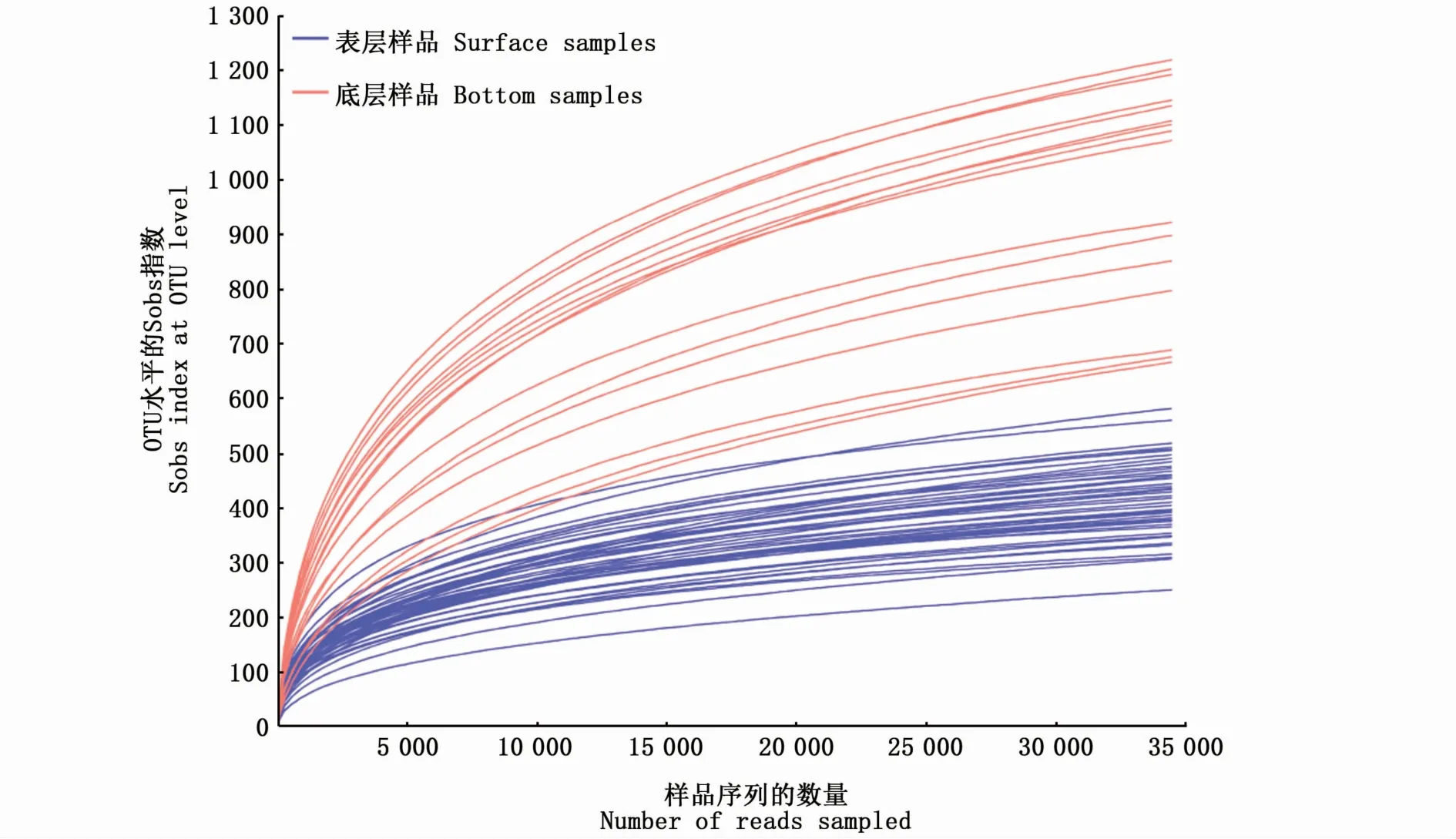
图2 稀释曲线Fig.2 Rarefaction curves
本次采集样品SW指数变化范围为1.714~5.350,Simpson指数变化范围为0.014~0.430,Ace指数变化范围为412.100~1601.303,Chao 1指数变化范围为394.588~1601.056。Wilcoxon秩和检验(Wilcoxon rank-sum test)结果表明,调查海域底层样品细菌多样性指数显著高于表层样品(P<0.05);底层样品之间无显著差异;表层样品之间也无显著差异,但多样性高值采样站位通常出现在本次研究区域的东北部水域。
2.2 细菌群落组成分析
本研究采集样品鉴定OTU隶属于37门、101纲、243目、404科、753属。其中13个门和20个纲中的OTU相对丰度超过1%。变形菌门(Proteobacteria)、蓝细菌门(Cyanobacteria)、放线菌门(Actinobacteria)和拟杆菌门(Bacteroidota)是研究区域中4个具有数量优势的门(图3,图4),各类群的平均相对丰度分别为39.24%、29.67%、10.97%和10.80%。其他相对丰度较低、但在大部分样品中均会出现的门级分类单元包括Marinimicrobia_SAR406_clade(占本次鉴定序列总数2.41%)、SAR324_clade(Marine_group_B,1.95%)和绿弯菌门(Chloroflexi,1.05%)。

图3 研究区域水体样品门级水平的细菌组成Fig.3 Composition of bacteria at phylum level in samples from the study area

图4 研究区域数量优势细菌门类的相对丰度热图Fig.4 Relative abundance heat map of numerically dominant bacteria in the study area
本研究样品中变形菌门共鉴定1038个OTU,丰度最高值出现在11号站位表层(占站位样品OTU总数86.99%)。变形菌门序列数占11号站位的表层、42号站位的底层、34号站位的底层、41号站位的底层样品序列总数75%以上。本次鉴定的变形菌门OTU中,397个隶属于α-变形菌纲,635个隶属于γ-变形菌纲,6个隶属于unclassified_p_Proteobacteria。α-变形菌纲在11站位的表层(82.57%)和7站位的表层(56.52%)是最具数量优势的纲,γ-变形菌纲在34站位的底层(78.99%)和42站位的底层(79.25%)样品中是最具数量优势的纲。
蓝细菌门在本次各调查站位均有出现,此类群共鉴定30个OTU,相对丰度最高值出现在24号站位的表层样品中(占站位样品OTU总数75.88%)。c_Cyanobacteriia和 c_Sericytochromatia是蓝细菌门中最具数量优势的纲。拟杆菌门共鉴定388个OTU,分属c_Bacteroidia、c_Kapabacteria和c_Rhodothermia纲。c_Bacteroidia包含OTU数量最多,共含377个OTU,丰度最高值出现在12站位的表层样品中(43.58%)。放线菌门共鉴定144个OTU,分属c_Actinobacteria、c_Acidimicrobiia、c_Coriobacteriia、c_Rubrobacteria、c_Thermoleophilia和unclassified_p_Actinobacteriota纲,丰度最高值出现在38站位的底层样品中(32.24%)。

表1 各采样站位OTU数量(97%相似水平)和多样性指数Tab.1 OTU number(97% similarity level)and typical diversity index of sampling stations
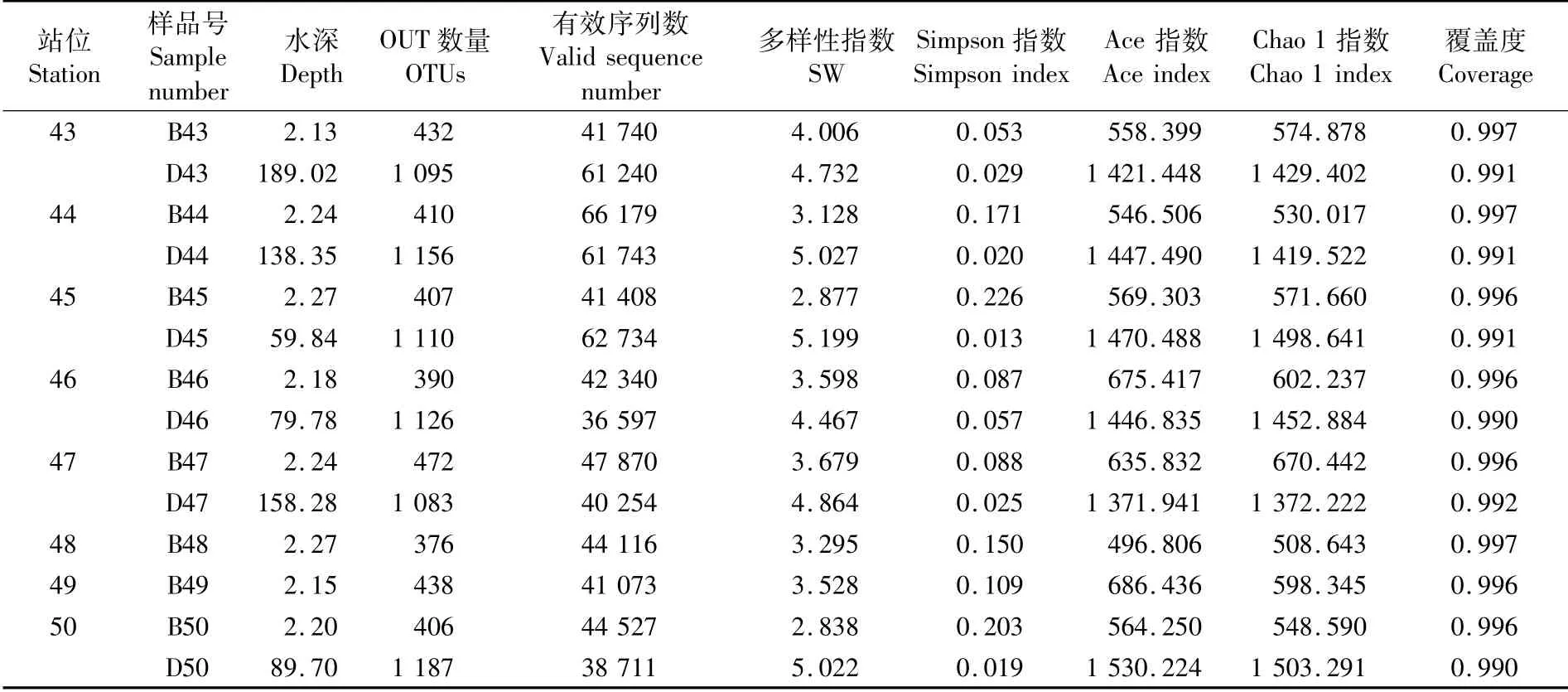
·续表1·
2.3 细菌群落β-多样性分析
NMDS和层级聚类分析表明,表层或底层海水中不同样品细菌群落之间差异较小,表层和底层海水之间细菌的群落结构差异较大(图5,图6)。ANOSIM分析结果显示,表、底层海水样品间细菌群落结构差异达到极显著性水平(R=0.6403,P=0.001)。
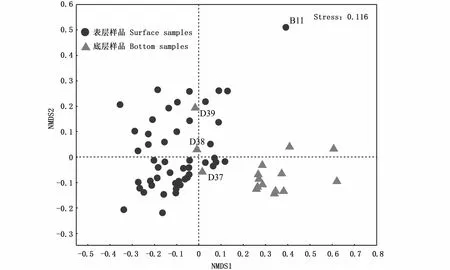
图5 海水细菌群落结构NMDS分析Fig.5 NMDS analysis of bacterial community in water samples
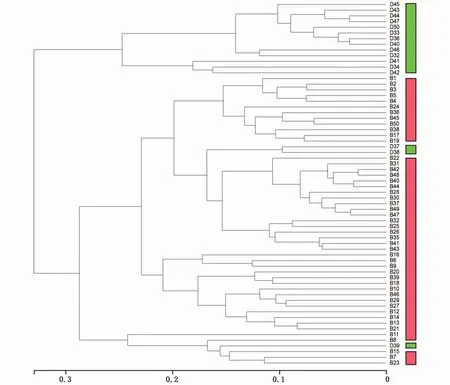
图6 海水样品细菌群落的层级聚类分析Fig.6 Hierarchical cluster analysis of bacterial community of seawater samples
2.4 不同组间的细菌物种差异组成
如图7所示,利用LEfSe分析工具的线性判别结果表明,典型地理区域内表层水体细菌群落的特征类群较为丰富。设定LDA值>3,北部湾水域表层特征细菌类群包括g_Cyanobium_PCC_6307、f_Halieaceae和o_Cellvibrionales等类群,琼东水域表层特征细菌类群包括 o_Rhodospirillales、g_norank_f_AEGEAN_169_marine_group和f_AEGEAN_169_marine_group,南海东北部表层水域特征细菌类群为o_Rhizobiales。相对于表层水体,底层水体样品中细菌群落特征类群更为丰富,特征细菌类群可达26个,包括g_norank_f_norank_o_norank_p_SAR324_cladeMarine_group_B、f_norank_o_norank_c_norank_p_SAR324_cladeMarine_group_B和c_norank_p_SAR324_cladeMarine_group_B等类群。

图7 南海水域中细菌群落的线性判别分析(仅显示LDA值>3的细菌类群)Fig.7 Linear discriminant analysis of bacterial community in South China Sea(only bacterial group with LDA>3 were listed)
3 讨论
3.1 南海水体浮游细菌群落多样性
本研究发现,底层海水样品中的细菌丰富度和多样性要高于表层样品,表现为层化现象,与全球层化模式较为相似[22]。已有针对南海浮游细菌群落研究报道的OTU数量多为数百个,例如YI等[23]调查南海海水中细菌垂直分布时报道表层细菌OTU数目范围为696~886;张益[10]在琼东5 m水层中发现浮游细菌OTU数量大多低于800个;南海范围内沿14°等纬度线断面5 m水层中得到的OTU数目为319~595[10]。本研究鉴定OTU数目范围为241~1245,OTU数量较大,变化范围较广,显示了南海浮游细菌较高的生物多样性。
3.2 南海水体的细菌组成
本研究结果表明,南海水体中最具数量优势的细菌类群包括变形菌门、蓝细菌门、放线菌门和拟杆菌门,此4类群分别占采集样品序列总数39.24%、29.67%、10.97%和10.80%。本研究揭示的南海水体中细菌群落优势类群组成与已有相关报道一致[24-25]。
变形菌门细菌在南海水体环境中不仅出现频率较高,而且通常具有较高的丰度。在变形菌门内,α-变形菌纲和γ-变形菌纲是本研究采集样品细菌群落的主要组成成分。在表层海水中,α-变形菌纲的相对丰度大于γ-变形菌纲;在底层海水中,γ-变形菌纲相对丰度大于α-变形菌纲。此种分布特征在已有相关研究中曾有提及[5,24],显示了同类群海洋细菌具多样化的环境适应能力。在目级分类水平,本研究样品中的优势类群与已有报道结论类似,例如α-变形菌纲主要包括红杆菌目(Rhodobacterales)、SAR11类群、红螺菌目(Rhodospirillales)、根瘤菌目(Rhizobiales)和鞘脂单胞菌目(Sphingomonadales),γ-变形菌纲主要包括交替单胞菌目(Alteromonadales)、Cellvibrionales、SAR86类群、Thiomicrospirales和假单胞菌目(Pseudomonadales)。此类数量优势类群的相对丰度在不同研究中存在差异,显示类群具有异质性的时空分布特点。例如红杆菌目相对丰度在细菌群落中的占比在大西洋和太平洋海水中大于25%[24],在南海北部少数站位中为5.44%[24],在本研究中为9.52%。
蓝细菌门相对丰度在本次采样的表层海水样品中占据显著数量优势,在24号采样站位的表层海水中的占比高达75.88%。底层海水中蓝细菌门的相对丰度较低。蓝细菌门细菌在海洋生态系统的碳循环和初级生产力形成中具有重要作用,并且为海洋生物提供碳源,本研究结论表明,蓝细菌门在南海水域生态系统物质循环和能量流动中发挥重要作用。已有研究表明,原绿球藻属、聚球藻属蓝细菌具有环境指示功能;原绿球藻属广泛分布在贫营养状态海域,聚球藻属喜居于营养丰富的水体环境[26-28]。在本次研究水域内,原绿球藻属在北部湾海域丰度极低,而在南海北部的陆架开阔海域表层水体(例如36号站位)丰度较高;聚球藻属在北部湾和南海北部沿海地区表层海水(例如24号站位)中丰度较高。蓝细菌的此种空间分布特征表明,北部湾和南海北部沿海地区的营养盐含量高于南海北部陆架开阔海域。
本研究区域内细菌群落结构可能与少数环境因子密切相关。例如,Marinimicrobia_SAR406_clade(Marine group A)和SAR324类群(Marine group B)是海水细菌中的重要类群,SAR406和SAR324类群在部分海水样品(第43号站位)中的相对丰度分别高达12.64%和10.12%。此2类群丰度在同站位表、底层水体中存在较为显著的差异,底层海水样品中的相对丰度较高。研究表明,SAR406和SAR324类群在南海、大西洋、太平洋海水垂直方向上的丰度差异与水体中溶解氧含量相关,水体溶解氧含量伴随深度增加而减少,此2类群在低氧区内具较高丰度[29-31]。
3.3 南海水体细菌的分布特征
细菌是南海生态系统中最具数量优势的生物类群,但是至今针对此区域内细菌多样性、空间分布特征以及群落和环境因子相关性等方面的认知仍较为有限。本研究采样范围较广,可提供较为少见的海盆尺度的空间分布特征分析。
本研究结果表明,南海表层或底层样品之间细菌群落多样性指数无显著差异(Wilcoxon秩和检验,P>0.05),样品间细菌群落结构较为相似(UPGMA聚类方法分析),区域内浮游细菌群落总体上未呈现出显著的空间异质性。尽管如此,细菌群落的多样性高值区通常出现在本次研究区域的东北部水域。此种差异应与东北部水域的环境特征相关。东北部采样水域在经度上位于珠江河口的两侧,在纬度上覆盖浅海至近千米深海,区域具有较高的环境异质性。珠江按流量是我国第二大河流,年径流量超过3492亿m3。珠江巨大的淡水输入在河口区域形成较为明显的盐度梯度。浅海和深海与陆地距离存在显著差异,陆源营养物质对区域的影响存在差别。此外,南海海洋动力过程具有季节性的多尺度变化特征,南海东北部上升流、海洋锋面、中尺度和次中尺度涡旋均可能影响海洋细菌分布格局及其多样性[32-33]。
南海水域细菌的垂直分布特征暂无较为明确的结论。部分研究表明,南海光合作用带范围内的细菌多样性和丰富度随水体深度的增加而增加[31],全球范围尺度上也是如此[22]。有研究认为,细菌在65 m水深处丰度最大,此区域通常也是叶绿素a含量的高值区[33]。海洋水文动力复杂,鱼类粪便、上升流引起的营养物质输入也可能在较深的水体形成细菌高值区[33-35]。本研究结果显示,南海水域细菌的垂直分布显示出显著的异质性。首先,底层水体样品中出现特异性细菌类群。Nitrospinota类群在南海北部底层海水样品中较为常见,在各采样站位中的相对丰度维持在较高水平(0.18%~8.82%,最低值和最高值分别出现在第38号和第47号站位)。KITZINGER等[36]利用酶联荧光原位杂交技术在表层海水中未发现Nitrospinota,而在表层以下不同水层均发现一定丰度Nitrospinota,并且随着海水深度逐渐增加直至到沉积物之上。Nitrospinota是海洋中最丰富的亚硝酸盐氧化细菌,是海洋中氮循环的重要参与者[37]。少数研究表明,Nitrospinota在黑暗海洋中固定15%~45%的碳[37]。其次,采样区域底层样品中细菌群落多样性显著高于表层(Wilcoxon秩和检验,P<0.05)。本次调查Shannon指数位于前10位的样品均采集于底层水体,其中33号站位底层水体细菌群落的Shannon指数最高。此外,部分OTU在不同水层中丰度存在显著差异。SAR11clade等在表、底层皆有分布的类群在底层水体具有更高的丰度。
南海是个开阔的海洋,受到诸如黑潮、季风、环流、上升流、涡流等物理因素以及珠江口等河口输入的影响,具有复杂的物理化学环境。本研究显示的底层水体细菌群落的较高多样性很可能与上升流影响有关。南海北部陆架海域主要的上升流区分别为闽南沿岸上升流、台湾浅滩上升流、粤东上升流、粤西上升流和琼东沿岸上升流。上升流将高营养盐的低温深层水带至真光层,能够改变上层海洋生态系统的微生物群落结构[38]。在上升流区域,CO2浓度的增加、营养物质的输入等情况能够显著提高该区域的初级生产力[39-40]。上升流区域底层水体中的细菌密度、生物量和细胞更大[34-35]。汪彧等[41]针对琼东海域上升流的观测研究指出,区域内强上升流影响区域主要包括海南岛东岸离岸100 km以内的30~50 m水体,此区域与本次研究的部分采样水域重合(43~50号站位),较为直接地证明了上升流对区域细菌多样性的促进作用。
