原发性骨髓纤维化向AML转化过程中造血干细胞差异表达基因的生物学功能分析及核心基因筛选
2021-12-18王艳张宇卉胡耐博滕广帅周圆白洁
王艳,张宇卉,胡耐博,滕广帅,周圆,白洁
1天津医科大学第二医院血液内科,天津300211;2中国医学科院血液病医院
(中国医学科学院血液学研究所)血液内科
原发性骨髓纤维化(PMF)是骨髓增殖性肿瘤(MPN)的一种常见类型,其发病机制与造血干细胞(HSC)的克隆性增殖关系密切[1],主要表现为脾大、全血细胞进行性减少、骨髓纤维化等。研究[2]显示,大约20%的PMF患者在确诊后的10年内会转化为急性髓系白血病(AML),预后极差。然而,PMF的发病机制错综复杂,其向AML转化的分子机制目前尚不完全清楚,明确PMF不良预后的分子机制可以为PMF的治疗提供新的理论依据。随着基因芯片技术的发展,基因测序已成为探索疾病分子机制的重要工具[3]。单细胞RNA测序可以检测每个细胞的RNA表达谱,与传统的基因芯片技术相比具有更高的分辨率[4]。单细胞测序技术可以检测到那些传统基因芯片技术难以检测到的稀有细胞基因,而这些基因可能具有及其重要生物学意义。本研究通过对GEO数据库中单细胞测序数据集GSE153319中的HSC进行生物信息学分析,筛选出PMF向AML转化过程中HSC的差异表达基因及核心基因,为探索PMF预后不良的分子机制及PMF的治疗提供新思路。
1 资料与方法
1.1 数据集的选取以“Primary myelofibrosis and Acute myeloid leukemia”为检索关键词,从GEO数据库中检索符合条件的单细胞测序数据集,最终选取基因芯片GSE153319为研究对象。GSE153319数据集包含1例PMF患者进展为AML过程中三个不同时间点(PMF慢性期、芦可替尼治疗后8个月、AML转化期)的样本。
1.2 HSC细胞和高变基因的筛选 利用R语言的Seurat包筛选GSE153319数据集中基因数量200~10 000和线粒体基因占比<5%的细胞,共获得符合条件的细胞7 717个。为了去除纳入研究细胞的批次效应,且最大程度保留细胞的基因表达信息,我们对7 717个细胞进行主成分分析(PCA)和T分布随机邻接嵌入(t-SNE)聚类,结果显示,7 717个细胞被划分为13个细胞簇,利用Seurat包的FindAllMarkers函数寻找每个细胞簇的特异性基因,并利用SingleR包和既往发表文献中的细胞标记基因对不同的细胞簇进行注释,最后利用VlnPlot函数和FeaturePlot函数显示HSC标记基因在不同细胞簇的表达,最终筛选出HSC。利用R语言对HSC进行PCA降维并可视化其主成分分布,筛选出HSC细胞间高度变化的基因(简称高变基因),选取MALAT1、VIM、HLADRA、FOS、JUN、CD74、TSC22D3、TMSB4X、MTRNR2、KLF6等排名前2 000的高变基因用于后续分析。
1.3 PMF向AML转化过程中HSC差异表达基因的筛选及基因本体(GO)功能富集和京都基因与基因组百科全书(KEGG)信号通路分析 利用R语言的Seurat包Findmarkers函数,设置筛选条件为|log2FC|>0.5且P<0.01,筛选PMF慢性期和AML转化期HSC的差异表达基因。利用DAVID(https://david.ncifcrf.gov)在线数据库 和Metascape(https://metascape.org)在线数据库对PMF向AML转化过程中HSC差异表达基因进行GO功能富集和KEGG信号通路分析,其中GO功能富集包括生物学过程(BP)、细胞组分(CC)、分子功能(MF)。
1.4 PMF向AML转化过程中HSC蛋白互作(PPI)网络图的构建及核心基因筛选借助STRING10(http://www.string-db.org)在线数据库构建差异表达基因的PPI网络图,并利用Cytoscape软件的Mcode插件筛选出核心基因簇,通过DAVID数据库对筛选出的核心基因簇进行GO功能富集,利用CytoHubba插件设置筛选条件为MCC算法【MCC(v)=∑c∈s(v)(|C|-1)!】,筛选出排名前5的基因,即为PMF向AML转化过程中HSC的核心基因。
2 结果
2.1 PMF向AML转化过程中HSC差异表达基因筛选结果 共筛选出98个差异表达基因,与PMF慢性期相比,AML转化期HSC中有78个上调基因、20个下调基因。上调的基因为FKBP5、RPL3P2、DUSP6、PNMT、ISG20、ARL4C、KLF7、FOS、KLF13、BTG2、RGS2、OSBPL10、MAL、JUNB、AL158827.2、MAN2A1、TIPARP、EGR3、TNFAIP3、TP53INP1、C5orf30、CXCR4、IER2、RHOB、MCL1、SAP30、AP002982.1、RPL5P17、LMNA、TXNIP、SOCS1、RPS2P55、AGPS、MTRNR2L10、AC062028.2、AKR1C2、STK17B、ID1、CD69、MMP7、AC009362.1、DUSP2、PER1、PTGS2、ID3、ZFP36、AC020916.1、GSTM3、CXCL8、EGR1、AREG、AC099340.1、GNAI1、RPL27AP、AC113367.1、SESN1、AC114760.2、NRXN2、RGS1、HIF3A、AKR1C1、ATP2B1-AS1、RASGEF1B、AL031733.2、SOCS3、S100A10、KLF2、MYADM、AL356512.1、AL445433.1、TSC22D3、KLF6、KLF4、ARRDC3、WASF4P、PIK3IP1、KLF9、ZNF595。下调的基因为IFI44L、PABPC1P4、PARP9、STAT2、IFIT3、PTAFR、RPS26P47、IFITM1、CLU、STON2、STAT1、TRIM69、TNFSF13B、IFI6、AC095059.2、DTX3L、HLA-H、OAS1、IRF7、MX1。
2.2 差异表达基因的GO功能富集和KEGG信号通路分析结果 GO功能富集分析结果显示,上调基因的BP主要富集在RNA聚合酶Ⅱ启动子转录的负调控、炎症反应的负调控、成纤维细胞生长因子反应、成骨细胞分化的负调控以及血管新生等,CC主要富集在细胞核和细胞质,MF主要富集在DNA结合以及RNA聚合酶Ⅱ核心启动子的转录激活子活性;下调基因的BP主要富集在炎症反应、干扰素-γ介导的信号通路以及细胞对α干扰素的反应,CC主要富集在细胞质。KEGG信号通路分析结果显示,上调基因的KEGG信号通路主要富集在TNF信号通路、癌症相关通路、凋亡相关通路以及Apelin信号通路等。
2.3 PMF转化为AML过程中HSC核心基因的筛选结果 构建的PMF向AML转化过程中HSC差异表达基因编码的PPI网络图见图1。利用Cytoscape的Mcode插件分析获得2个核心基因簇,基因簇1主要与免疫调控、JAK-STAT信号通路、成纤维细胞生长因子反应以及细胞增殖等关系密切,基因簇2主要与DNA转录以及凋亡调控关系密切。根据MCC算法筛选出排名前5的核心基因分别是FOS、EGR1、PTGS2、CXCL8和CXCR4,这5个基因可能是PMF转化为AML过程中HSC的核心基因。
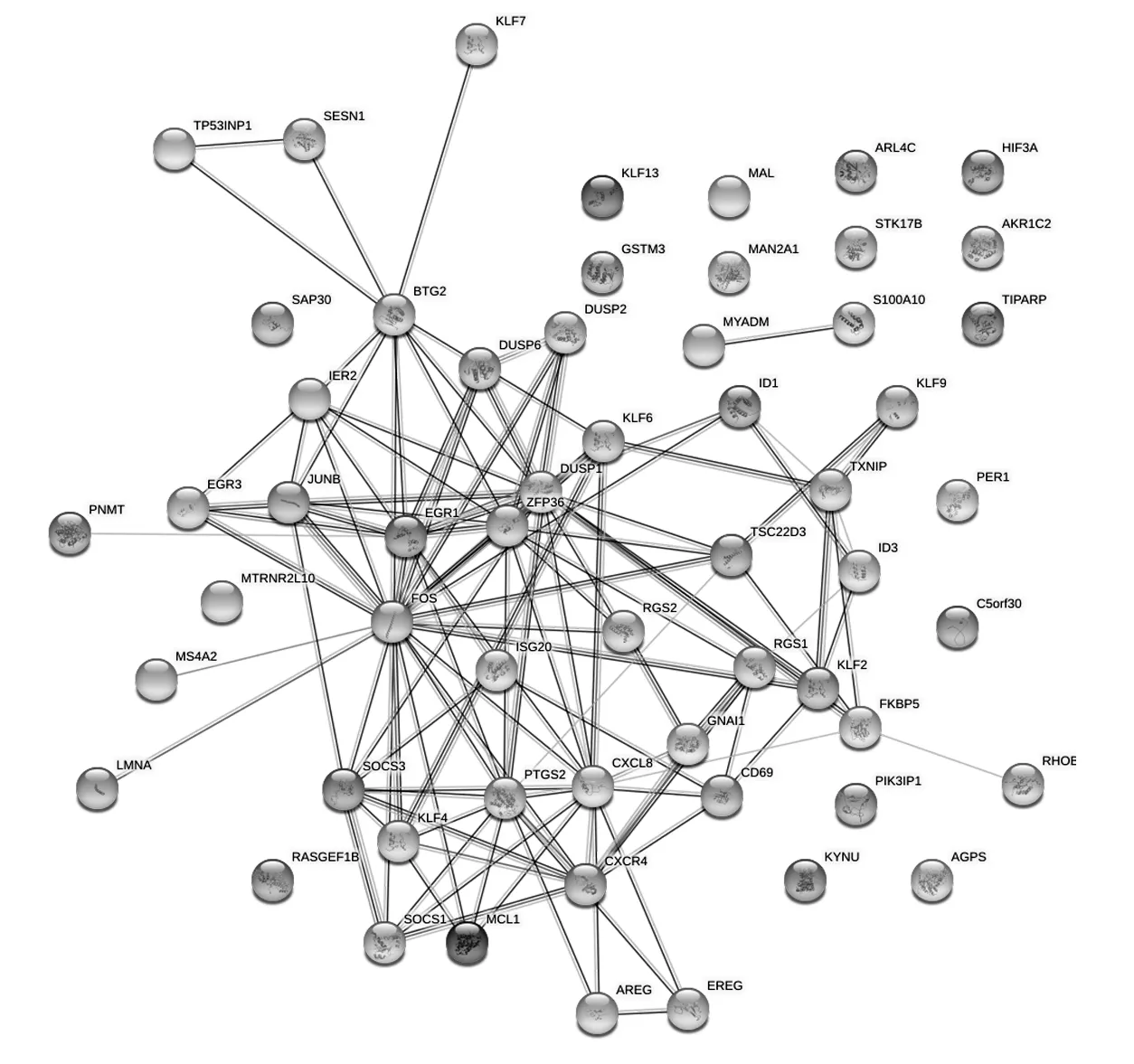
图1 PMF向AML转化过程中HSC差异表达基因编码的PPI网络图
3 讨论
PMF是一种罕见而具侵袭性的MPN,其致病因素为HSC的克隆增殖紊乱,主要表现为全血细胞减少、髓外造血和全身症状。目前PMF的治疗方式主要为常规对症治疗、靶向药物治疗和HSC移植[5]。常规治疗主要通过糖皮质激素、雄激素治疗骨髓纤维化相关性贫血[6],对于脾大的骨髓纤维化患者可应用羟基脲[1]。靶向药物治疗主要是指JAK抑制剂在PMF患者中的应用,主要包括芦可替尼、菲卓替尼等。随着技术的发展,靶向药物的研究也取得了突破性进展,从而使得PMF患者的生存期和生存治疗都有了较大提升[7-8]。HSC移植是患者接受放化疗或联合免疫抑制剂清除体内肿瘤细胞后,通过回输HSC以重建造血系统和免疫系统的一种治疗方式,是目前唯一可能治愈PMF的方法[9]。然而,由于PMF本身的异质性以及移植后并发症的存在,使得HSC移植的应用存在一定局限性。目前PMF的治疗仍存在很大难度,且一旦转化为AML,预后极差。本课题组通过检索GEO数据库中与PMF不良预后有关的单细胞数据集,利用生物信息学技术,寻找PMF进展为AML的关键生物标志物,为PMF的治疗提供新的理论依据。
本课题组通过R语言的Seurat包、SingleR包以及既往文献将单细胞数据集GSE153319中的7 717个细胞划分为13个细胞簇。既往研究[10]发现,HSC的克隆增殖紊乱是PMF的主要发病机制。因此,本研究通过R语言提取HSC进行差异表达分析,筛选PMF预后不良的关键分子机制。通过对差异表达基因进行GO功能富集发现,PMF进展为AML的差异表达基因主要富集在炎症调控、细胞增殖以及分化等过程中,KEGG信号通路则主要富集在TNF信号通路、癌症相关通路、凋亡相关通路以及Apelin信号通路等,提示PMF进展为AML的过程中,HSC的增殖、分化功能以及对骨髓微环境免疫调控的功能发生了改变。通过Cytoscape进一步筛选出2个核心基因簇,其中基因簇1主要与免疫调控、JAK-STAT信号通路、成纤维细胞生长因子反应以及细胞增殖等关系密切,基因簇2主要DNA转录以及凋亡调控关系密切,进一步验证了HSC增殖、分化紊乱以及免疫调控在PMF不良预后中的作用。
利用Cytoscape的Cytohubba插件根据MCC算法筛选出排名前5的核心基因,分别是FOS、EGR1、PTGS2、CXCL8和CXCR4。研究[11-12]显示,FOS和EGR1属于原癌基因,可以协同作用,共同促进细胞的增殖、分化,在AML的发生发展中发挥重要作用。PTGS2可促进细胞的增殖、分化,在AML患者中的表达水平明显升高,其表达水平与肿瘤预后密切相关[13]。CXCL8是趋化因子的一种,可以促进HSC的增殖、分化,与AML的发生发展关系密切[14]。CXCR4是趋化因子CXCL12的特异受体,在调控细胞增殖、髓外移行、浸润、黏附及对化疗药物的耐药中起重要作用[15-16]。ABDELOUAHAB等[17]学者发现,骨髓纤维化患者的CXCR4通路被过度激活,是PMF诊断的敏感标记物。
综上所述,本研究通过检索GEO数据库中PMF不良预后的单细胞测序数据集,对PMF和AML样本中的HSC进行生物信息学分析,确定与PMF不良预后相关的差异表达基因。与PMF慢性期相比,AML转化期HSC中有98个差异表达基因;差异表达基因主要富集在炎症调控、细胞增殖以及分化等过程中,参与TNF信号通路、癌症相关通路、凋亡相关通路以及Apelin信号通路等;FOS、EGR1、PTGS2、CXCL8、CXCR4等5个差异表达基因可能是PMF转化为AML过程中HSC的核心基因。本研究通过分析预测了PMF预后不良的可能发病机制并筛选出参与PMF进展为AML的核心基因,为PMF的诊治提供了新的思路。然而,本研究还具有一定的局限性,本研究完全基于对GEO数据库中的数据进行生物信息学分析,对筛选出的核心基因仍需通过实验进行进一步验证。
