砷剂调控肿瘤细胞命运的机制及激活抗肿瘤免疫的潜力
2021-12-17陈锦锋马瑜婷
陈锦锋 马瑜婷
砷剂治疗疾病在中国已有逾2000年的历史,近年来,其在肿瘤治疗领域的应用受到广泛关注。砷剂与细胞中多种生物分子的巯基以及蛋白质中的半胱氨酸残基有很高的亲和力,而这些蛋白大多参与胞内氧化还原状态的调节。因此,砷剂暴露会导致胞内活性氧(reactive oxygen species,ROS)的生成和累积,进而触发多种应激反应和死亡方式,包括氧化应激、自噬、凋亡、程序性坏死、焦亡、铁死亡等[1-5]。抗氧化剂N-乙酰-L-半胱氨酸能阻断三氧化二砷(arsenic trioxide,ATO)诱导的细胞死亡,提示ROS 是关键触发因素[6]。细胞死亡过程能引发肿瘤抗原的暴露,并伴随着一系列“危险信号”分子(又称cell death-associated molecular patterns,CDAMPs)的释放,引发肿瘤细胞“抗原性”与“佐剂原性”的双重提升,有助于增强肿瘤细胞的“免疫原性”[7]。目前,ATO 诱导的细胞死亡是否能影响肿瘤细胞的免疫原性尚未明确。本文主要总结ATO 的临床应用现状和发展瓶颈,分析ATO 可能诱导的多种细胞死亡方式,及其相互调控的分子机制,并就ATO 用于肿瘤免疫治疗的新策略及可行性进行探讨和展望。
1 砷剂治疗肿瘤的概况
上世纪70年代以来,张亭栋、马军、陈竺、王振义等国内多位医学专家先后在ATO 治疗急性早幼粒细胞白血病(acute promyelocytic leukemia,APL)的疗效和机制探索中获得重大突破。多项国内外研究证实,ATO 对原发性和复发性APL 患者的完全缓解率(complete response,CR)可达85%~90%,且5年生存率较高[8]。鉴于其良好的临床治疗效果,ATO 相继被美国食品药品监督管理局、欧洲药品管理局批准用于APL 的治疗[9]。
ATO 在APL 治疗上的成功为其他类型血液肿瘤的治疗提供了理论依据。研究发现,ATO 与全反式维甲酸联合可治疗携带IDH2 突变的急性髓细胞性白血病(acute myeloid leukemia,AML )(约占AML 患者的20%)[10]。对于FLT3-ITD 突变型AML(约占AML 患者的25%),ATO 能诱导内质网应激等促进肿瘤细胞凋亡,改善患者预后[11]。此外,对慢性髓细胞白血病、复发和获得性耐药的多发性骨髓瘤、成人T细胞白血病和骨髓增生异常综合症患者,ATO 也具有治疗潜力[12-15]。值得注意的是,APL 对ATO 有较高的敏感性,低剂量药物(0.15 mg·kg-1/d)便可有效治疗患者,而大多数非APL 血液肿瘤需要较高剂量的药物(0.25 mg·kg-1/d),其临床应用受限于严重的不良反应(表1,ClinicalTrail.gov 网站已公布的部分血液肿瘤临床试验)。

表1 ATO 治疗血液肿瘤的临床试验
ATO 能有效控制晚期原发性肝癌肺转移,延长患者总体生存期[16]。然而,对于其他大多数实体肿瘤(如肺癌、食道癌、胃癌、大肠癌、胰腺癌、膀胱癌、子宫癌、乳腺癌、口腔癌等),其临床应用非常有限。可能原因包括:1)药物难以到达并留存于肿瘤病灶;2)药物在体内代谢失活并排出;3)药物损伤正常组织和免疫系统,产生较强的不良反应。为改善ATO对实体瘤的疗效,研究者尝试了多种联合策略。遗憾的是,ATO 与放疗、化疗、小分子靶向药物等联用虽然能增强其胞毒作用,但临床疗效的改善并不显著。近年来,脂质体、聚合物胶束、多孔硅等纳米载体递送策略极大地提高了ATO 的生物利用度,改善了其体内药代动力学,提高了靶向性,降低了不良反应[17]。然而,ATO 纳米药物也面临着一些技术瓶颈,包括:载药量低、缓释量常难以满足治疗需求;易被单核吞噬细胞系统等清除;病灶局部药物聚积可抑制免疫细胞浸润。因此,探索合适的剂量、给药途径、药物联合策略是ATO 用于实体瘤治疗的重要命题(表2,ClinicalTrail.gov 网站已公布的部分实体肿瘤临床试验)。
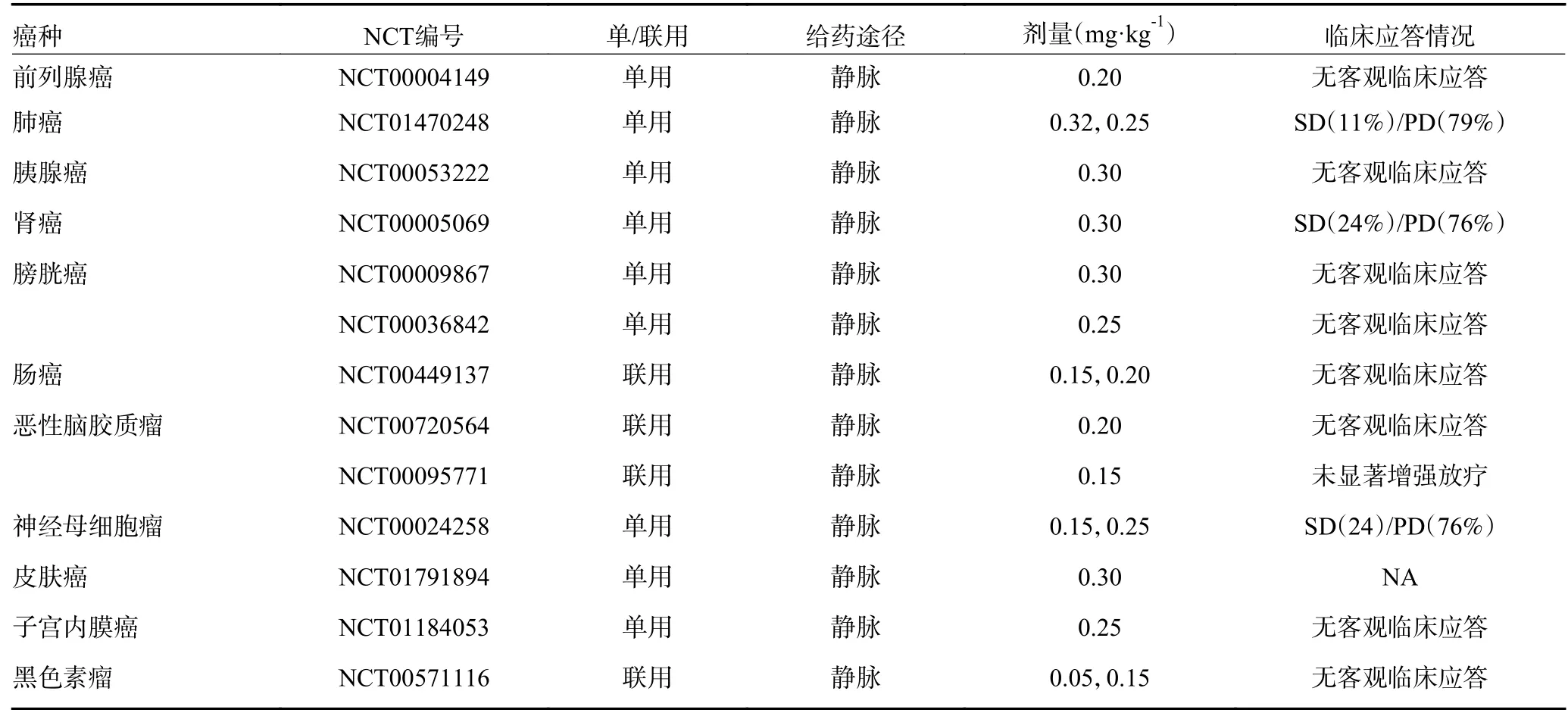
表2 ATO 治疗实体肿瘤的临床试验
2 砷剂调控肿瘤细胞命运的分子机制
ATO 作用下的肿瘤细胞命运如何,不同细胞命运的关键调控机制如何,是值得深入探索的重要科学问题。ATO 可直接或间接作用于细胞内诸多分子及信号通路,影响多种细胞应激和死亡方式,包括:氧化应激、内质网应激、自噬、凋亡、程序性坏死、焦亡、铁死亡等。图1 显示细胞应激反应与死亡方式的关键调控机制和形态学特征:凋亡细胞膜起泡、DNA 浓缩以及断裂,形成凋亡小体并释放内容物;自噬过程形成自噬小体,随后与溶酶体结合成自噬溶酶体对隔离底物进行降解;程序性坏死中磷酸化的MLKL 能插入细胞膜形成孔道,膜破裂,该过程常伴随ROS 的增加;焦亡主要依靠CASP 的激活,并活化GSDM 蛋白转位到膜上,形成孔洞,胞质外流;铁死亡表现为线粒体变小、脊变少、脂质过氧化等,该过程受GPX4、ASCL4等调控。

图1 细胞应激反应与死亡方式的关键调控机制和形态学特征
研究发现,ATO 引发的氧化应激会扰乱细胞内蛋白折叠,错误折叠蛋白累积于内质网中可诱导内质网应激,触发未折叠蛋白反应、内质网相关蛋白质降解等以恢复细胞稳态。持续或过度的内质网应激可促进ROS 生成,引发恶性循环并导致细胞凋亡[18]。ATO 可破坏线粒体膜的通透性,释放细胞色素C 并与APAF1、半胱氨酸蛋白酶9(CASP9)结合形成凋亡小体,招募并活化CASP3/6/7 等,对胞内蛋白进行高效切割,诱导细胞以CASP 依赖的方式凋亡[19]。此外,ATO 能显著增加BCL-2 家族中促凋亡蛋白(如BAX、BAK)的表达及其向线粒体的转运[20]。ATO 诱导的ROS 还能直接开启线粒体膜通透性转换孔,导致凋亡诱导因子的释放及入核,诱导核固缩及染色质碎片化,触发非CASP 依赖的凋亡[21]。
自噬是细胞在外界不良刺激下维持内环境稳态的重要机制。自噬溶酶体可降解自身蛋白或受损细胞器,以便循环使用代谢产物。研究表明,ATO 可增强自噬相关蛋白LC3A/B、Beclin1 的表达,激活JNK、p38-AMPK 通路,并抑制PI3K/Akt-mTOR 通路,从而引发自噬。ATO 联合卡莫司汀可以提高ROS/GSH比例,诱导实体瘤细胞发生自噬性死亡[22]。ATO 处理人成骨肉瘤细胞,可导致转录因子TFEB 在Ser142 位点去磷酸化并入核,继而促进自噬溶酶体必需蛋白的表达,加速自噬性死亡[6]。自噬对肿瘤进展的影响具有双面性,一方面,自噬可缓解内质网应激与氧化应激,减少DNA 损伤和基因突变,限制细胞的癌变及快速增殖,阻碍肿瘤的早期发生;另一方面,自噬可帮助癌细胞适应恶劣的肿瘤微环境,促进其存活和进展。而在肿瘤治疗过程中,自噬一方面有助于癌细胞产生药物抵抗,另一方面也能参与诱导自噬样细胞死亡(由自噬分子机制驱动的非凋亡、非坏死的细胞死亡形式)。
焦亡是一种程序性的细胞炎性坏死,依赖于CASP1/4/5/11、颗粒酶等的活化,以及其对Gasdermin(GSDM)蛋白家族分子的切割。GSDM 的N 端结构域能在细胞膜上寡聚成孔,引发细胞裂解性死亡并促进白介素-1 细胞因子家族成员的成熟与释放。无机砷可诱导β 细胞焦亡和炎症小体的活化[2],而ATO 处理胶质瘤细胞U251 后能激活CASP3 和GSDME 依赖的焦亡和乳酸脱氢酶的释放[3]。
铁死亡是一种依赖于二价铁离子、由酯氧合酶介导的新型细胞死亡形式。ROS 等自由基在胞内聚集、以谷胱甘肽过氧化物酶GPX4 为核心的抗氧化系统的耗竭,最终可导致脂膜上的不饱和脂肪酸发生脂质过氧化,并造成细胞膜破裂。ATO 可诱导大量ROS 生成,提示ATO 可能和铁死亡密切相关。最新研究证实,无机砷可诱导胰岛细胞MIN6 发生铁死亡,且该过程可以被铁死亡小分子抑制剂ferropstatin-1阻断[4]。
程序性坏死是一种受丝氨酸/苏氨酸蛋白激酶RIPK1 和RIPK3 精密控制的、CASP 非依赖性的细胞死亡。由RIPK1 和RIPK3 形成的坏死小体可导致MLKL 的寡聚化和磷酸化,最终产生细胞膜损伤、胞质肿胀、细胞器膨大、释放细胞内容物并引发炎症反应。目前尚缺乏ATO 能诱导细胞程序性坏死的直接报道,但有研究表明,ROS 能通过直接氧化RIPK1上的三个关键半胱氨酸,增强其自身磷酸化,促进程序性坏死的发生[5]。
在不同的细胞死亡命运的调控中,CASP8 或许是关键的分子开关。研究发现,抑制CASP8 可导致TNFα 诱导的细胞死亡由凋亡转变为程序性坏死。程序性坏死途径受阻时,丧失酶活性的CASP8 可充当蛋白支架,促进炎性小体复合物形成并导致细胞焦亡[23],而自噬导致的CASP8 降解能促使凋亡转变为程序性坏死[24]。值得注意的是,本研究组前期研究提示,不同的细胞死亡形式可同时存在于肿瘤组织中。可能原因为:肿瘤组织的细胞构成复杂(肿瘤细胞、基质细胞、多种免疫细胞);肿瘤细胞克隆存在高度的异质性;组织内不同区域的微环境有差异;细胞死亡逐渐进行而并非同步发生。此外,就单个细胞而言,不同的细胞死亡途径能否同时或先后激活,不同细胞死亡途径的关键执行分子能否借助相分离等机制富集在细胞内不同区域,值得细致观察、深入探索。
3 砷剂诱导肿瘤发生免疫原性细胞死亡
细胞应激及死亡可打破组织局部稳态,引发免疫应答和炎性反应。细胞死亡如何被免疫系统有效感知,不同细胞死亡形式引发免疫应答的类型是否相同,均是值得深入探索的问题。传统观点认为凋亡可诱导免疫耐受,尽管具体机制尚不够明确,但凋亡过程中CASP3/6/7 的激活可以下调固有免疫应答的多个关键调控因子(如cGAS、MAVS、IRF3 等)[25]。细胞坏死过程中伴随着细胞内容物的大量释放,有学者认为这些物质属于细胞固有成分,而非诱导产生的“危险信号”,虽然能诱导强烈的炎症反应,但并不一定能激活抗肿瘤免疫应答[26]。肿瘤细胞的“免疫原性”是指癌细胞被免疫系统识别和清除的特性,由“抗原性”及“佐剂原性”共同决定。濒死的肿瘤细胞能释放肿瘤抗原,并释放或暴露多种损伤相关分子模式(DAMPs),包括钙网蛋白(CALR)、高迁移率族蛋白B1(HMGB1)、腺苷三磷酸(ATP)、I 型干扰素(IFNα/β)、膜联蛋白A1(ANXA1)等。上述“危险信号”分子可调控抗肿瘤免疫应答,也成为公认的免疫原性细胞死亡的生物标志物[7]。图2 显示ATO 用于治疗型疫苗制备的策略:ATO 介导的癌细胞死亡不仅暴露肿瘤抗原,同时能释放一系列“危险信号”分子(如ATP、CALR、F-actin、IFNα/β、HMGB1、AnexA1),这些信号分子能与单核细胞、髓系树突状细胞(DC)细胞上的相关受体结合,促进抗原吞噬和提呈,并激活T 细胞,最终抑制肿瘤生长。
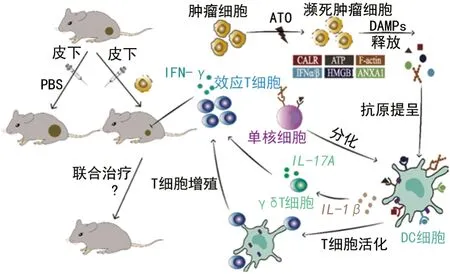
图2 ATO 用于治疗型疫苗制备的策略
该领域研究的关键科学问题包括:不同细胞死亡形式对肿瘤细胞“免疫原性”的影响、对上述“危险信号”分子释放的调控机制。多项研究表明,凋亡并非意味着免疫耐受,蒽环类药物诱导肿瘤细胞凋亡可促进CALR 和磷脂酰丝氨酸暴露于胞膜表面,增强其抗原的摄取和提呈,提升其免疫原性。借助泛CASP 抑制剂Z-VAD-fmk 不可逆地阻断细胞凋亡,虽然不影响肿瘤细胞的死亡,但其免疫原性显著下降[27]。濒死细胞释放ATP 依赖于凋亡和自噬通路,多种CASP 分子、自噬相关基因(autophagy related genes,ATG)都参与了这一过程。ATG5 和ATG7 缺陷型肿瘤细胞的自噬通路受阻,ATP 释放量显著减少,化疗效果大幅降低[28]。ATP 的膜外释放也受到CASP 蛋白的调控,CASP3 可切割缝隙连接蛋白家族成员PANX1,后者活化后形成跨膜通道并导致ATP 的释放,而ZVAD-fmk 可以有效阻止该过程[28]。程序性坏死通路的活化有助于ATP 和HMGB1 的释放,从而增强肿瘤细胞的免疫原性。RIPK3 或MLKL 缺陷型肿瘤对化疗不敏感,借助三磷酸腺苷双磷酸酶抑制剂提高组织内ATP 浓度,联合使用TLR4 激动剂能显著增强上述缺陷型肿瘤细胞的化疗效果[29]。目前,尚不明确铁死亡是否参与调控肿瘤细胞的免疫原性,特别是“危险信号”分子的释放和暴露,有文献提示HMGB1 可能是铁死亡相关的DAMPs[30]。
4 砷剂对免疫细胞的直接作用
除了诱导肿瘤细胞死亡,砷剂可直接作用于免疫细胞,引发免疫功能障碍。砷剂可改变免疫细胞DNA 甲基化水平,导致基因表达谱重编程,影响细胞的脂质代谢、pH 调节、NF-κB 信号转导等[31]。低浓度的ATO 能诱导单核细胞坏死,减少DC 的产生,降低DC 的吞噬、抗原提呈和细胞因子分泌功能[32]。巨噬细胞是参与抗原呈递和清除非己成分的专职吞噬细胞。体外实验提示,ATO 可严重阻碍单核细胞向巨噬细胞分化,并通过NF-κB 途径抑制巨噬细胞的黏附能力和吞噬活性[33]。砷的积累还能抑制淋巴细胞增殖、迁移和活化,诱导T 细胞凋亡,并通过增加氧化应激和减少IL-2 的分泌来抑制T 细胞活性,这也是慢性砷中毒导致鲍文病的主要原因[34]。ATO 对免疫细胞的抑制效应也被运用于治疗自身免疫疾病,如改善骨髓移植造成的排斥反应和缓解系统性红斑狼疮[35-36]。值得注意的是,ATO 在某些情况下还可以作为佐剂增强免疫应答。ATO 能增强自然杀伤细胞(NK)对APL 的杀伤活性[37]。低剂量的ATO 能诱导氧化应激和亚硝化应激,选择性地清除调节性T 细胞(Treg),增强效应T 细胞对结肠癌和乳腺癌的杀伤活性[38]。骨髓来源的抑制性细胞(MDSC)对建立及维持免疫耐受、免疫抑制至关重要,ATO 可诱导MDSC 凋亡或促进其分化,该发现有助于拓展探索ATO 临床应用的新可能[39]。
5 结语与展望
传统的砷剂治疗主要通过静脉滴注的方式,虽然有部分口服制剂被批准用于临床治疗,但目前应用较少。虽然砷剂在体外实验中具有广谱的抗癌活性,然而其应用于实体肿瘤的临床治疗进展有限,仅在原发性晚期肝癌治疗中显示出一定效果。究其原因,主要有如下几点:1)ATO 是水溶性化合物,容易全身扩散,难以富集至肿瘤病灶部位。由于肝脏是重要的解毒器官,ATO 相对富集,局部浓度相对较高,更易诱导肝癌细胞死亡;2)ATO 的全身扩散容易导致免疫细胞死亡,引发系统性免疫抑制;3)由于ATO 具有较强的不良反应,体内用药的剂量受限,难以达到有效杀伤肿瘤细胞的浓度;4)由于ATO 的高水溶性,纳米载体材料包裹ATO 并靶向递送的策略虽然有应用前景,但严重受限于低下的药物包封率。因此,探索砷剂治疗实体瘤的新策略有望带来重要临床突破。
部分化疗药物预处理的肿瘤细胞能用作预防型疫苗,接种至小鼠皮下可有效抑制肿瘤的再活化。对于肿瘤患者而言,治疗型疫苗的应用前景远大于预防型疫苗。本团队新开展的预实验提示,实体瘤细胞在ATO 处理后能出现多种细胞应激反应和死亡方式,可释放多种“危险信号”分子,具备增强肿瘤细胞免疫原性的潜力。因此,借助ATO 在体外预处理自体来源的肿瘤细胞,可制备治疗型全细胞肿瘤疫苗。该策略具有如下优势:可有效避免药物在体内的不良反应;可广泛覆盖肿瘤细胞表达的多种抗原;可选择最佳治疗窗口多次注射;可根据特定肿瘤类型优化药物浓度和处理时间等条件,最大限度地诱导免疫原性细胞死亡,通过激活持久的抗肿瘤免疫降低肿瘤的复发和转移。
上述治疗型疫苗可与其它多种治疗策略联用,旨在实现协同抗癌效应。近些年来,基于免疫检查点阻断的免疫疗法取得了巨大的突破,但治疗应答率亟需提升,新型不良反应的发生也带来新的挑战。肿瘤微环境中缺乏T 淋巴细胞浸润,也称为“冷肿瘤”,是导致PD-1 单抗疗效不佳的原因之一,而联合使用周期素依赖性激酶抑制剂(如dinaciclib)诱导免疫原性死亡,激活DC 并增加T 细胞浸润有望显著提升PD-1单抗的疗效[40]。基于ATO 的治疗型全细胞肿瘤疫苗与免疫检查点阻断联用,是否能发挥1+1>2 的效果值得尝试和探索,该策略对晚期肿瘤患者的临床获益,很有可能大于化疗。
