Transcriptome Sequencing and Expression Profiling of Pea in Leaf Color Mutant
2021-12-16ZHENGXingweiZHENGJunWANGJun
ZHENG Xingwei,ZHENG Jun,WANG Jun
(1.Institute of Wheat,Shanxi Agricultural University,Linfen 041000,China;2.Institute of Millet,Shanxi Agricultural University,Changzhi 046000)
Abstract:Understanding the molecular mechanisms of chloroplast development and chlorophyll biosynthesis in pea(Pisum sativum L.)is extremely important for pea breeding.This study used high-throughput transcriptome sequencing and bioinformatics analyses to examine changes in gene expression in leaves of yellow-leaf mutant(yl)and wild type(WT),and compared the gene expression profiles of these two pea types.A total of 25 599 903 and 24 806 761 high quality reads were obtained for the WT and the mutant,respectively.In total,4 423 differentially expressed genes(DEGs)were identified in yellow-leaf mutant compared to WT,of which 2 449 were up-regulated and 1 974 were down-regulated.Gene ontology and pathway enrichment analyses indicated that the DEGs were involved in various biosynthetic and metabolic pathways.Expression profiles of genes involving chlorophyll synthesis and photosynthetic pathways were illustrated.Quantitative RT-PCR analysis of the relevant DEGs confirmed the RNA-Seq results.Expression patterns of DEGs related with Chl metabolism showed that the key genes of chlorophyll synthesis and photosynthetic pathways may be related to the formation of yl.
Key words:Pisum sativum L.;ellow-leaf mutant;chlorophyll biosynthesis;transcriptome
Photosynthesis provides the energy necessary for green plants to maintain normal growth and development.The morphology,structure,number and internal chlorophyll(Chl)content of chloroplasts are important factors influencing the efficiency of photosynthesis.Mutations in genes related to photosynthesis can cause whitening[1],yellowing[2],light green coloration[3],streaks,spots[4],and other phenomena in green plants.Both natural mutations and artificially induced mutations may affect the development and functional integrity of plant chloroplasts,resulting in abnormal Chl content and a leaf color variation phenotype.
In the monocotyledonous rice and dicotyledonous soybean,researchers have used identified leaf color mutant materials to locate and clone a large number of leaf color mutant genes by forward and reverse genetics.There are currently 40 leaf color mutant genes cloned in rice,involving protoplast differentiation,Chl biosynthesis,photosynthesis membrane protein complex structure,fatty acid synthesis,nuclear signal transduction,bio-pigment anabolism and other pathways;the abnormal expression of regulatory genes involved in any of the links may lead to leaf color variation[5].More than 20 Chl-deficient mutant materials have been reported in soybean,and some Chl-deficient sites have been mapped by molecular marker technology.For example,the YL_PR350 gene located on chromosome 15 is involved in thylakoid membrane development[6].The GmCHLI1a/b gene encoding the magnesium chelatase subunit was mapped using the mutants y11-2,CD-5,y9,and cd1[7-8].
Pea(Pisum sativum L.)is an important protein-rich legume crop globally,and it was the original model organism used in Mendel's discovery of the laws of inheritance,making it the foundation of modern plant genetics.Due to the size(4.5 Gb)and repetitive nature of the pea genome,progress in pea genomics,especially for transcriptomic analyses,has lagged behind many other plant species.Most ofthe mutant studies have focused on seed development[9]and nitrogen fixation[10-11],and little is known about the molecular mechanisms of chloroplast development and chlorophyll biosynthesis in pea.In this study,comparative transcriptome profiles of the yellow-leaf mutant and wild type of pea were analyzed by RNA-Seq.Based on a combination of biochemical analysis and bioinformatics,we identified the major metabolic pathways related to leaf color for the loss of pigmentation in pea.
1 Materials and methods
1.1 Plant material
The pea cultivar Zhongwan 4 is widely used for commercial production in China and represents typical features of pea grains.One Chl-deficient mutant developed spontaneously in the field,with a mutation frequency of approximately 1.0×10-4-1.5×10-4.Leaf chlorophyll relative content was measured using a portable Chlorophyll meter(SPAD-502,Minolta,Japan).Stomatal conductance and photosynthetic parameters were detected using the LI-6400 portable pho tosynthesis system(LI-COR,Lincoln,Nebraska,USA)at 10:00 am.Three individual plants were assayed for each of the mutant and wild-type(WT)plants,and the fully expanded functional leaves were detected.
1.2 Methods
1.2.1 Sample collection and preparation For transcriptome analysis,yl and WT leaves were collected from Zhongwan 4 seedlings at 9:00 am,with three replicates for each type.The six samples were used to construct cDNA libraries designated yl1,yl2,yl3,WT1,WT2,and WT3.Samples were frozen in liquid nitrogen immediately and stored at-80 ℃until RNA extraction.Total RNA from different samples was extracted using the TRIzolReagent(Invitrogen Life Technologies,Shanghai,China).RNA purity was checked using a NanoPhotometerspectrophotometer(IMPLEN,CA,USA).RNA concentration was measured using the QubitRNA Assay Kit in a Qubit2.0 Fluorometer(Life Technologies,CA,USA).RNA integrity was assessed using the RNA Nano 6000 Assay Kit of the Agilent Bioanalyzer 2100 system(Agilent Technologies,CA,USA).The cDNA library construction and sequencing of six RNA samples were completed by Beijing Biomarker Technologies Co.,Ltd(Beijing,China).
1.2.2 Short reads de novo assembly and gene functional annotation First,clean reads were obtained by removing adapter and primer sequences,and low-quality reads from raw data.Then,de novo transcriptome assembly was carried out using the Trinity assembly program[12].Unigene function was annotated based on the following databases:NR(NCBI non-redundant protein sequences),Swiss-Prot(a protein sequence database with manual annotation and review),Pfam(Protein family),COG(Clusters of Orthologous Groups of pro teins),KOG(EuKaryotic Orthologous Groups),EggNOG(v4.5),KEGG(Kyoto Encyclopedia of Genes and Genomes),and GO(Gene Ontology).
1.2.3 Identification of differentially expressed genes(DEGs)Differential expression analysis of yl and WT(each with three biological replicates)was performed using the edgeR program package.Differential expression analysis between two samples was performed using the DEGseq(2010)R package.The P value was adjusted using q value[13].q value<0.005 and|log2FC|>1 was set as the threshold for significantly differential expres sion.GO enrichment analysis of the differentially expressed genes(DEGs)was implemented by the topGO R package[14]based on the Kolmogorov-Smirnov test.KOBAS[15]software was used to test the statistical enrichment of DEGs in KEGG pathways[16].
1.2.4 Validation by real-time quantitative PCR Total RNA used for qRT-PCR analysis was extracted from the leaves of the mutant and WT plants with three biological replicates using the same method as described above.cDNA synthesis and qRT-PCR were carried out according to the methods used in ZHENG et al[17].The relative expression level of each gene was calculated usingthe2-ΔΔCtmethod[18]and wasnormalized tothecontrol gene Actin.
2 Results and analysis
2.1 Analysis of photosynthetic parameters of Zhongwan 4 yellow leaf mutant and wild type
Tab.1 showed the Chl content,net photosynthetic rate,transpiration rate and stomatal conductance values of Zhongwan 4 yellow leaf mutant and WT.The results of analysis of variance showed that the photosynthetic parameters of mutants and WT were significantly different(P <0.01).The yellow leaf mutant had a low photosynthetic rate,low transpiration rate,and low stomatal conductance.
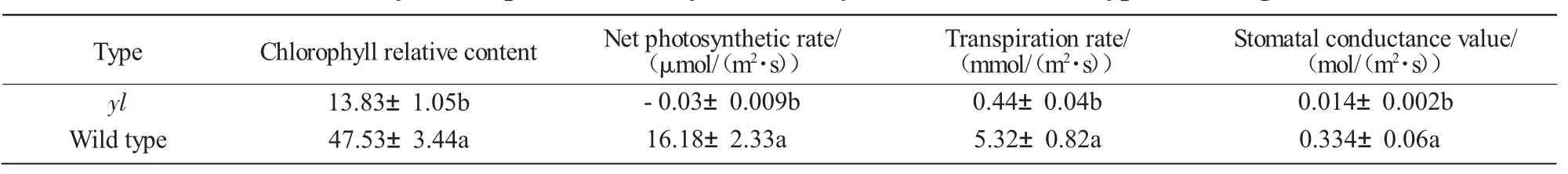
Tab.1 Photosynthetic parameters of yellow-leaf(yl)mutant and wild type of Zhongwan 4
2.2 RNA-seq and de novo assembly
We performed RNA-Seq analyses of leaves from yellow-leaf mutant and WT of Zhongwan 4 pea.After cleaning and quality checks,a total of 24 806 761 and 25 599 903 clean reads with 7 410 398 314 and 7 639 503 665 nucleotides were produced for yl and WT,respectively(Tab.2).Q30 percentages of the clean data ranged from 89.99%to 90.21%,meeting the requirements for further analysis.Due to lacking an appropriate reference genome sequence,all clean reads were assembled de novo with the Trinity method[12].Tab.2 results shows that more than 80% of the clean reads could be mapped back to the assembled transcripts,which resulted in a total of 97 682 unigenes with a mean length of 718.24 bp.The size distributions of these unigenes are in Fig.1.The statistical summary of data is outlined in Tab.2.
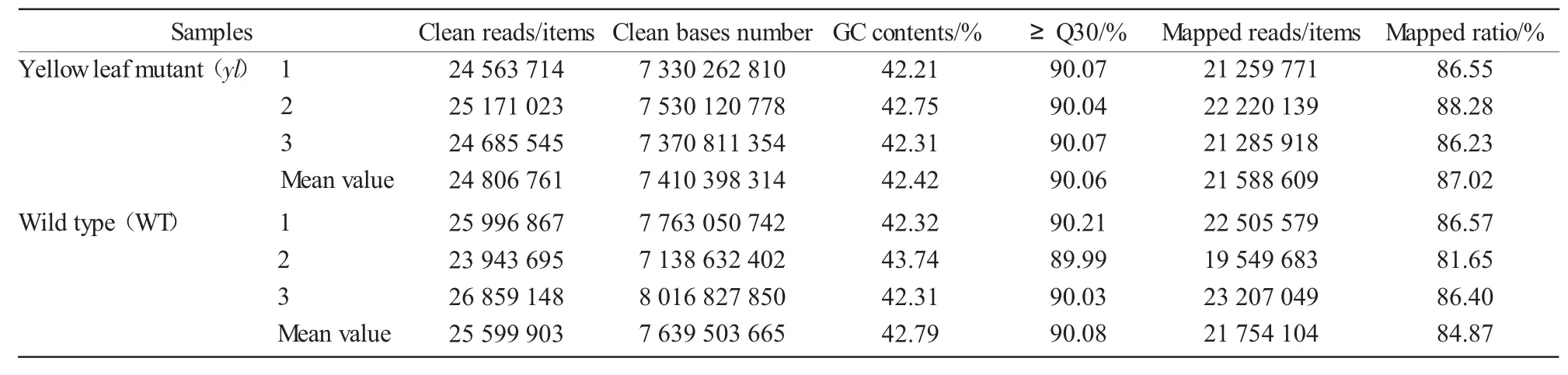
Tab.2 Total number of RNA-Seq data collected from yl mutant and wild type of Zhongwan 4
2.3 Functional annotation and structure of unigenes
Public databases(NR,Swiss-Prot,COG,KOG,EggNOG,Pfam,GO,and KEGG)were used to annotate all unigenes with local BLAST(E value<1.0E-5)and HMMER(E value<1.0E-10)programs.The results showed that 48 433(49.58%of 97 682)unigenes were matched to one or more of the databases(Tab.3).CDS prediction analysis was made using the command "get ORF" in the TransDecoder package.A total of 62 907(64.40%)CDS of unigenes were identified.Using microsatellite(MISA)identification tool,a total of 6 313 SSR were identified in unigenes longer than 1 kb.
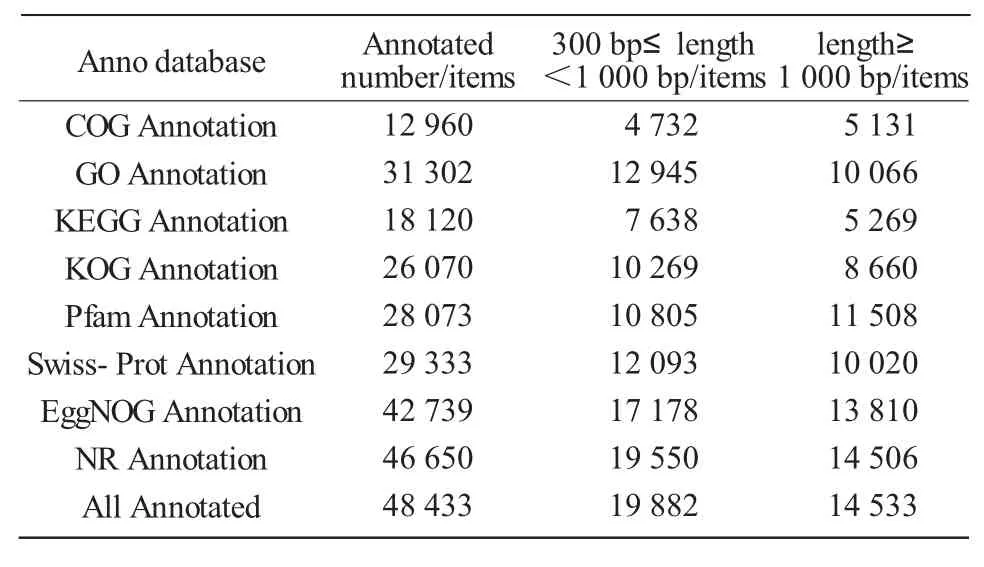
Tab.3 Gene annotation
2.4 Differentially expressed genes(DEGs)between yl and WT plants
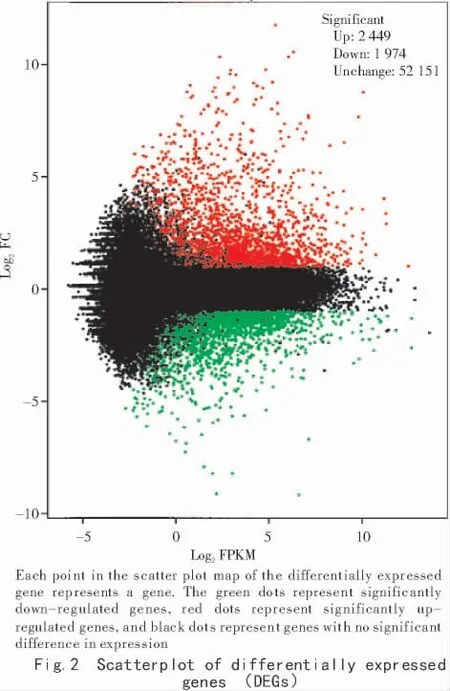
In order to find the differentially expressed genes(DEGs)in leaves of yl and WT,clean reads of these libraries were respectively assigned to all unigenes using RSEM(RNA-Seq by Expectation Maximization)software,and the differences in expression levels ofunigenes were calculated with the FPKM method.The DEGs were determined with a two-fold or greater change(|log2FC|>1)and false discovery rate(FDR)of 0.001 or less.The expression ratio(yl/WT)of the unigenes in two libraries varied greatly.Among them,only 4 423(4.53%)showed a two-fold or greater expression difference between yl and WT.Ofthese,1 559(35.24%)had expression ratios between 2 and 5,and a smaller percentage(5.67%)showed more than a five-fold difference in expression level between the two libraries(Fig.2).Compared with WT,the number of up-regulated and down-regulated DEGs in the yl samples was 2 449 and 1 974,respectively.
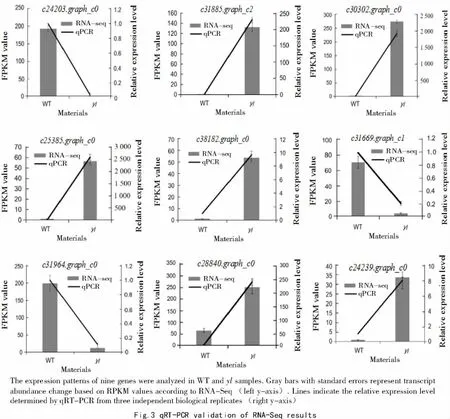
2.5 Validation of the differentially expressed genes(DEGs)via RT-qPCR
To confirm RNA-seq results,we performed qPCR assays with samples of identified DEGs.Nine genes were chosen based on their statistically significant up/down-regulated expression in the RNA-seq analysis.As shown in Fig.3,the expression patterns of genes between the two samples in the qRT-PCR and RNA-seq analyses generally followed the same trends,but the qRT-PCR method was more sensitive than microarray analysis for detecting transcript expression.
2.6 GO functional enrichment of DEGs
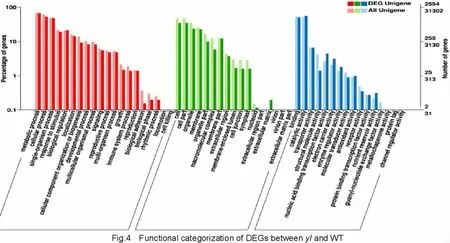
GO(gene ontology,http://www.geneontology.org/)is an international classification system for standardized gene functions with three categories:biological process,cellular component,and molecular function.The categorization of DEGs between yl and WT according to GO categories is shown in Fig.4.In the biological process(BP),2 091 genes(47.27%of the DEGs)were anno tated,with the largest number of genes involved in processes ofphosphorylation(GO:0016310)(261,12.48%),transcription(GO:0006351)(234,11.19%),regulation of transcription(GO:0006355)(204,9.76%),protein phosphorylation(GO:0006468)(170,8.13%),and DNA metabolic process(GO:0006259)(74,3.54%).In the molecular function(MF)category,2 666 genes(60.28%of the DEGs)were enriched in oxido reductase activity(GO:0016491)(348,13.05%),nucleic acid binding(GO:0003676)(320,12.00%),phosphorus-containing groupstransferaseactivity(GO:0016772)(294,11.03%),DNA binding(GO:0003677)(214,8.03%),and zinc ion binding(GO:0008270)(140,5.25%).With regard tocellular component(CC)category,a total of759 genes(17.16%)were significantly enriched,involving almost all cell components including chloroplasts,plastids,thylakoids,cell membranes,and cytoplasm.Most genes were enriched in the following components:integral component of membrane(GO:0016021)(268,35.31%),intracellular non-membrane-bounded organelle(GO:0043232)(85,11.20%),organelle subcompartment(GO:0031984)(42,5.53%),plastid thylakoid(GO:0031976)(41,5.40%),chloroplast thylakoid(GO:0009534)(41,5.40%),chloroplast thylakoid membrane(GO:0009535)(35,4.61%),and plastid thylakoid membrane(GO:0055035)(35,4.61%).
2.7 KEGG pathways of DEGs
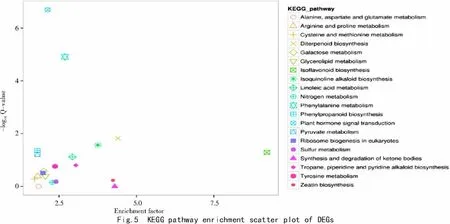
In organisms,different genes coordinate with each other to perform biological functions,and pathway annotation analysis of DEGs using the KEGG(Kyoto Encyclopedia of Genes and Genomes)database is helpful to further understand gene functions.According to the KEGG annotations,687 DEGs(15.53%)were annotated into 123 metabolic pathways in detail,and 32 were observed as significantly enriched(P-value≤0.05)for DEGs of yl vs WT.The top eight represented pathways were 'plant hormone signal transduction'(ko04075)(68),'phenylalanine metabolism'(ko00360)(34),'diterpenoid biosynthesis'(9),'isoquinoline alkaloid biosynthesis'(ko00950)(10),'phenylpropanoid biosynthesis'(ko00940)(37),'isoflavonoid biosynthesis'(ko00943)(4),'pyruvate metabolism'(ko00620)(35),and'linoleic acid metabolism'(ko00591)(12)(Fig.5).
2.8 Analysis of differentially expressed genes in chlorophyll synthesis and photosynthetic pathways
In this study,the yellow leaf mutant was found to have special photosynthetic characteristics.Exploring the photosynthesis metabolism genes in its transcriptome revealed the signaling pathway and regulation mechanism of yellow leaf mutant photosynthesis.Based on the functional annotation and classification of significant DEGs,we analyzed the related gene of Chl metabolism,carotenoid biosynthesis,photosynthesis,and carbon fixation in photosynthetic organisms.There are 36 unigenes involved in Chl metabolism and photosynthetic pathways.On this basis,a total of 15 highly differentially expressed genes were screened under the condition of |log2FC|>2,and a heat map was drawn(Fig.6).There were 11 up-regulated and 4 down-regulated of expression levels in these genes.
In the Chl degradation pathway,Chl is catalyzed by chlorophyllase to form detached chlorophyllide a,and the gene that regulates chlorophyllase was significantly up-regulated in the yellow leaf mutant.In the Chl synthesis pathway,the gene encoding the magnesium-protoporphyrin IX monomethyl ester cyclase(CDR1)was down-regulated in the mutant material.CDR1 and chlorophyllase are key enzymes that regulate Chl synthesis and degradation.Up-regulated expression of the gene encoding the chlorophyllase gene and the down-regulated expression of the CDR-encoding gene are closely related to the decrease in Chl of the mutant strain.

Chloroplasts are organelles for photosynthesis,and the synthesis of photosynthetic pigments is mainly carried out on the thylakoid membrane of chloroplasts.The genes encoding the photosystem II Psb P,Psb R and photosystems I Psa K,Psa L,Psa N,and the F-type ATP synthase b subunit located on the thylakoid membrane were down-regulated in the mutant.Photosystem II Psb P and F-ATPase were significantly down-regulated(Fig.6).In the photosynthetic biochar fixed metabolic pathway in the yellowleaf mutant,the expression of phosphoenolpyruvate carboxylase was downregulated,but almost all other enzymes showed up-regulated expression,including fructose-bisphosphate aldolase,transketolase,glyceraldehyde-3-phosphate dehydrogenase,fructose-1,6-bisphosphatase,phosphoenolpyruvate carboxykinase,NADP-dependent malic enzyme 4,and pyruvate phosphokinase(Fig.6).
3 Discussion
The formation of plant yellow leaf mutants is closely related to the content of Chl and carotenoids.The synthesis and degradation pathways ofChl in mature plants are highly coordinated processes catalyzed by a series of enzymes encoded by nuclear genes.Mutations in any of the enzyme genes cause a decrease in Chl content,which can lead to leaf color variation.The three genes CHLH,CHLD,and CHLI encode the three subunits ofmagnesiumchelatase.Yellowor yellow-green color mutants currently caused by mutations in the CHLH-encoding gene have been reported in Arabidopsis and rice[19-20],and Chl-deficient mutants associated with the CHLI gene are reported in soybean[21].In Arabidopsis and rice leaf color mutants,expression of the POR gene in the process of Chl synthesis significantlyreduced chlorophyllase[22-23].In this study,unigenes encoding magnesium chelatase and POR did not show significant up-regulation or down-regulation in yellow leaf mutants.The unigene encoding the magnesiumprotoporphyrin IX monomethyl ester cyclase(CDR1)was down-regulated in the mutant material,indicating that the same leaf color mutation trait may be caused by different gene mutations.There were fewreports of mutations in the leaf color caused by mutations in the magnesium-protoporphyrin IX monomethyl ester cyclase,therefore,it is speculated that this unigene may be a newgene that causes a Chl deletion mutation.
The gene encoding chlorophyllase was significantly up-regulated in mutant materials,and chlorophyllase was the first enzyme associated with Chl degradation to be cloned,so earlier studies suggested that the regulation of the degradation pathway enzyme begins with chlorophyllase.Later,some researchers questioned this because in Arabidopsis the single knockout and double knockout chlorophyllase encoding genes AtCLH1 and AtCLH2 did not prevent chlorophyll degradation during senescence.Researchers considered pheophorbide an oxygenase(PaO)an essential enzyme in Chl degradation.Although chlorophyllase plays a key role in Chl degradation,studies have shown that chlorophyllase also functions directly in fruit ripening[24]and postharvest Chl degradation[25].
