聚合氮化碳的制备、改性及光催化还原二氧化碳性能研究
2021-12-15李佳慧李克艳宋春山郭新闻
李佳慧,李克艳,宋春山,2,郭新闻
(1.大连理工大学化工学院,辽宁大连116024;2.香港中文大学理学院化学系)
伴随着工业化的进程,化石燃料的大量燃烧使得大气中二氧化碳(CO2)浓度急剧增加,导致温室效应和生态环境恶化,制约了人类社会的可持续发展。为了缓解上述问题,可对CO2进行资源化利用,即对产生的CO2进行捕集、提纯,然后转化为高附加值的化学品和燃料。常用的技术方法有生物转化法[1]、催化加氢法[2]、电催 化还原法[3]和光催化还原法[4]等。其中,光催化还原CO2技术是以半导体材料为光催化剂,利用光照下激发出的光生电子对CO2分子进行还原。此方法实现了太阳能向化学能的转化,符合可持续发展的战略要求,是一种极具吸引力的CO2资源化途径。
1 光催化还原CO2的反应机理
CO2分子非常稳定,C=O键能为750 kJ/mol,因此打破化学键对其进行还原相当困难。根据特定反应途径和转移的电子数,CO2还原反应可产生许多不同的产物,常见的产物有甲酸(HCOOH)、一氧化碳(CO)、甲醛(HCHO)、甲醇(CH3OH)、甲烷(CH4),或 者 乙 烷(C2H6)、乙 烯(C2H4)、草 酸(C2H2O4)、乙 醛(CH3CHO)、乙醇(C2H5OH)、高级醇等高级碳氢化合物[5],见表1。由表1可知,不同的产物具有不同的还原电位,很少有半导体能提供足够的电位将单个光生电子转移到游离的CO2分子上,反应实质上遵循质子辅助的多电子转移步骤[6]。目前大部分对CO2光还原的研究集中在CO2还原反应上,氧化反应则是通过提供空穴牺牲剂完成[7]。

表1 CO2还原反应的标准电势[6]Table 1 Standard potentials for CO2 reduction reactions[6]
光催化还原CO2反应的实验条件多为含牺牲剂、助催化剂或光敏剂的有机液相体系[8],或是需要加热或加压的水相及气相体系[9-10]。光照条件多为可见光或者含部分紫外光的模拟太阳光[11-13]。光催化还原CO2反应机理涉及到电子和质子的转移、氢化和脱氧过程、C—O键的断裂和C—H键的生成等[14]。反应机理复杂且产物多样,产物的选择性与热力学因素(光子能量、导带位置)和动力学因素(光强、活性位点、电荷分离、反应物和中间体的吸附与解吸)密切相关[15]。反应中间体可以是分子、阴离子或自由基,其形成过程和稳定性很大程度上取决于催化剂的表面性质,且两者的相互作用影响着反应途径和产物分布[16]。反应过程中还需要H2O或H2等作为提供氢的还原剂,而选择不同的还原剂同样会导致反应路径出现差异[7],与此同时竞争反应不仅存在于H2O的还原和CO2还原反应中,而且还存在于由不同反应路径产生的不同还原产物中[15]。
因此,针对某一还原产物可能会存在多种反应途径的解释,结合密度泛函理论计算、原位傅里叶变换红外光谱或电子顺磁共振(EPR)等表征手段对中间体的检测,可探究某一特定光催化剂在给定实验条件下的反应途径。TERRANOVA等[17]研究表明,*COOH中间体的形成是光催化剂SrTiO3生成CO反应的速控步骤。YU等[18]制备了Cl掺杂的Cu2O纳米棒,发现*COOH、*CO和*CH3O中间体的生成有助于高选择性生成CH4。PANG等[19]合成了含Zn空位(VZn)的ZnS,VZn的存在有助于COO-*的形成并进一步转化为HCOO*,从而提升了HCOOH的产量。WANG等[20]发现,PCN中掺杂S可降低生成*COOH和*CH3O中间体的自由能,从而提高CH3OH的产量。PARK等[21]制备了Cu、CdS量子点修饰的钛酸钠纳米管,铜颗粒的存在有利于CH3·中间体的形成,并通过CH3·—CH3·偶联反应生成了C2+产物。由此可见,光催化还原CO2生成某种产物的反应机理与催化材料的性质和反应条件有关。
2 聚合氮化碳(PCN)
由于CO2分子超高的稳定性,开发高活性的光催化剂是实现其高效转化的关键。光催化材料的选择需要平衡两个对立面:一方面,为了具有较高的太阳光利用效率,材料需要具备可见光吸收能力,这就要求半导体有相对较小的带隙;另一方面,带隙又必须跨越与光催化反应相关的还原和氧化电位的范围[22],对于CO2光还原反应来说,半导体的导带位置要高于CO2还原反应电位。聚合氮化碳(PCN)具有可见光响应、易于合成、能带位置合适等优势,近年来成为光催化还原CO2领域的研究热点[23]。
2.1 PCN的几何结构
图1为PCN两种可能存在的化学结构,即以三嗪环(C3N3)为结构单元和以七嗪环(C6N7)为结构单元。通过密度泛函理论计算以及热力学分析发现,以七嗪环为结构单元的PCN更加稳定[24]。石墨烯的结构为sp2-杂化碳组成的二维薄片,PCN与之有相似之处,为sp2-杂化的碳和氮原子组成的π共轭平面。FINA等[25]通过粉末X射线衍射和中子衍射研究了PCN的三维结构,由于相邻层中π电子之间存在斥力,七嗪环组成的各层之间存在错位堆叠。另外,通常在525℃以上热解尿素、硫脲、单氰氨、双氰氨、三聚氰胺以及其他含碳、氮的前驱物得到的黄橙色粉末并不是石墨相氮化碳,而是叫melon的聚合氮化碳[26]。亚胺单元将七嗪环单元连接形成一维Z字型的链,不同的链之间通过氢键形成二维结构。前驱体的不完全聚合使结构中存在大量氨基基团,形成了无定形或半晶态的相,导致光催化性能不佳[27]。若能克服聚合过程中的动力学问题,有效地减少缺陷和末端氨基的存在,可生成高结晶性的PCN以提高光催化性能。

图1 PCN的(a)三嗪环和(b)七嗪环结构单元[11]Fig.1 Triazine(a)and tri-s-triazine(b)structure units of PCN[11]
PCN的结构通常用X射线衍射(XRD)和傅里叶变换红外光谱(FT-IR)进行表征。通常情况下,在XRD谱图中,2θ为27.4°处的峰代表PCN的(002)晶面,来自PCN的层间堆叠,2θ为13.1°处的峰代表PCN的(100)晶面,来自七嗪环基本结构单元的面内堆叠。FT-IR图中,3 000~3 400 cm-1处的峰来自于末端氨基的振动,1 200~1 700 cm-1和810 cm-1处的峰分别为七嗪环的伸缩振动和呼吸振动的信号。
2.2 PCN的电子结构
PCN内部的自由电子浓度远大于空穴浓度,是一种n型半导体,见图2。由图2可知,PCN具备中等的带隙(约为2.7 eV)以及合适的导带(CB,-1.3 V)与价带(VB,+1.4 V)位置,其导带电子具有足够的还原能力,可将CO2转化为HCOOH、CO、CH3OH、CH4等产物。
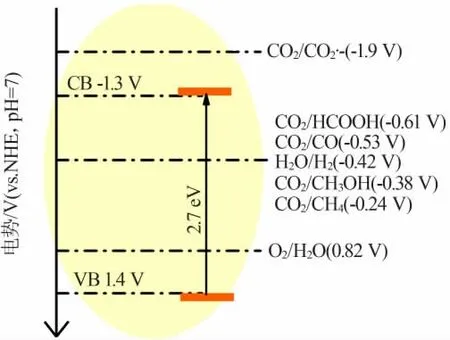
图2 PCN的能带位置示意图[28]Fig.2 Energy band positions of PCN[28]
3 PCN的制备
PCN的制备方法分为物理法和化学法。其中物理法包括离子注入法、反应溅射法、激光束溅射法等[29];化学法包括热聚合法、溶剂热法、超分子自组装法、电化学沉积法等,相对于物理法,化学法对设备要求低,更简便易行,是目前常用的方法。
3.1 热聚合法
在空气或惰性气体氛围、常压、温度为500~600℃条件下直接热解尿素、单氰胺、双氰胺、三聚氰胺、硫脲或密胺等富氮前驱体2~4 h就可得到黄色的产物[30]。制备过程为加聚和缩聚反应的结合,图3是以氰胺为前驱体制备PCN反应途径。具体步骤:在203℃和234℃下,中间体氰胺缩合成双氰胺和三聚氰胺;在335℃左右可发现含三聚氰胺的产物;进一步加热到390℃,三聚氰胺重排形成七嗪环;最后,在520℃左右发生进一步缩合,生成PCN;600℃以上时PCN变得不稳定;超过700℃时将发生分解[31]。
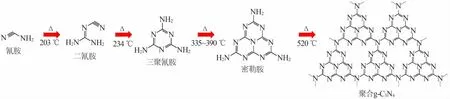
图3 以氰胺为前驱体制备PCN的反应途径[31]Fig.3 Reaction pathway for the preparation of PCN using cyanamide as the precursor[31]
以尿素为前驱体制得的PCN产率较低,而以三聚氰胺为前驱体时产率较高。但直接热解法制得的PCN存在比表面积小的缺点,使其催化性能受到一定限制[32]。利用模板法可制备高比表面积、大孔径的PCN。硬模板法的主要步骤:1)浸渍前驱体于模板中;2)高温焙烧模板孔道内的前驱体,使其缩聚生成PCN;3)使用HF或NH4HF2对模板进行溶除。罗磊[33]提出了择形沉积的制备策略,以MCM-41为模板,三聚氰胺为前驱体,制备出了高比表面积的薄壁单壳层和双壳层PCN。传统硬模板法制备中空或多壳层材料依赖于前驱体对模板剂孔道的填充,而此法是将三聚氰胺沉积聚合于模板表面。具体的步骤:三聚氰胺首先迁移进入MCM-41的孔道,并随着温度的升高转变为低聚物,当温度高于450℃时,MCM-41孔道的产物由于择形效应使得碳、氮迁移出“狭窄”的孔道,并沉积聚合在更加开放的外表面,形成壳层结构。
3.2 溶剂热法
溶剂热法由水热法发展而来,选择适当溶剂,将前驱体放入高压釜中于高温高压条件下进行反应。反应体系均匀、过程容易控制,且可防止有毒副产物的挥发。WANG等[34]以三聚氯氰和双氰胺为前驱体,乙腈为溶剂,在高压釜中180℃反应24~96 h可得氧掺杂中空微米球结构的PCN。XIA等[35]以盐酸胍和双氰胺为前驱体、乙腈为溶剂,在溶剂热条件下于180℃反应48 h得到了富含氰基和羧基缺陷的高结晶性片状PCN。
3.3 超分子自组装法
超分子自组装法是通过前驱物分子间的弱相互作用力,如三聚氰胺与三嗪衍生物之间的氢键相互作用,形成有序的超分子组装体,再进一步焙烧热聚合制备PCN。通过控制自组装过程的反应参数和条件,可以有效地调控PCN的组成、结构、形貌、光电性质,从而提高PCN的光催化性能[36]。GU等[37]将三聚氰胺和三聚氯氰分散在乙腈中,在高压釜中于200℃溶剂热反应24 h,得到的前驱物在550℃氩气流中煅烧2 h获得多孔PCN微球。YU等[38]将三聚氰胺、三聚氰酸和草酸在二甲基亚砜溶剂中进行超分子自组装,得到的固体前驱体在550℃氮气氛围中煅烧4 h制得三维花环状的PCN。
3.4 电化学沉积法
通过外加电流作用于一定浓度的含氮前驱体电解液,可使前驱体发生一系列的氧化还原反应生成PCN薄膜。此法设备简单、容易控制、不需要高温高压。BAI等[39]以丙酮为溶剂、二氰二胺为前驱体,采用电化学沉积法合成了直径为0.8~1.1μm、厚度为0.08~0.25μm由5~30 nm颗粒组成的结构为空心球的PCN。
4 PCN的改性及光还原CO2性能的提升
4.1 形貌调控
目前研究者们已经制备出纳米片[40]、纳米球[41]、空心微球[42]、一维[43]以及介孔[44]等多种形貌的PCN。FU等[45]首先对三聚氰胺进行一步热聚合,之后在600℃温度下N2气流中对其进行二次煅烧,通过热剥离纳米片的过程,成功地制备出了PCN纳米管(记为CN-Tube,图4a),相较于一步热聚合得到的块状氮化碳(记为BCN,图4b),PCN纳米管的比表面积增大,光吸收性能变好,光生载流子分离效率提高。相较于BCN,CN-Tube光还原CO2产生CH3OH的速率由0.17μmol/(g·h)提升至0.88μmol/(g·h)。TANG等[46]以三聚氰胺和NH4Cl为前驱物,制得了三维气泡结构的PCN。其中NH4Cl作为气体模板,加热分解产生的NH3和HCl气体削弱了纳米片层之间的π-π相互作用,形成了三维气泡结构的超薄氮化碳(CN-U),堆叠的纳米片厚度为1.6 nm,远远低于块状BCN的厚度(10.5 nm)。CN-U拥有更高的比表面积,有利于活性位点的暴露和光生载流子的传递,相较于块状BCN,其光还原CO2生成CH4的速率提高了3.16倍。

图4 CN-Tube(a)和BCN(b)的SEM照片[45]Fig.4 SEM images of CN-Tube(a)and BCN(b)[45]
4.2 异原子掺杂
在PCN中掺入其他元素可在其能带中形成杂质能级,并作为光生载流子的捕获中心,促进电荷分离,延长载流子的寿命,拓宽光催化剂的光响应范围。光催化性能的提升取决于掺杂元素的种类和掺杂量,过量的掺杂有可能成为载流子的复合中心,反而使得光催化性能变差[47]。
4.2.1 非金属掺杂
氧、硫、磷、卤素等原子可以掺杂进入PCN骨架调控其电子结构。WANG等[48]通过在520℃下煅烧硫脲制备出硫掺杂的TCN,相较于由相同制备条件下的三聚氰胺制得的MCN,其带隙由2.70 eV减小为2.63 eV。由于TCN中存在杂质S的能级,故而光生电子很容易从杂质态跃迁到导带,或者从价带跃迁到杂质态。在模拟太阳光的照射下,在水相中反应3 h后TCN和MCN光还原CO2生成CH3OH的产量分别为1.12、0.81μmol/g。
4.2.2 金属掺杂
多种金属掺杂进入PCN可实现光催化性能的提升,例如Fe[49-50]、Cu[51-52]、K[53-54]和Zn[55-56]等。 较低含量的金属掺杂可引入局部电子态,在光催化剂价带的上方形成施主能级或导带的下方形成受主能级,从而缩小半导体的带隙。而足够高的掺杂量会在带隙中间形成离域能带,实现半导体的多光子激发,但需注意只有具有合适能带位置的中间能隙态才具有足够强大的氧化还原电位,这样在改善催化剂光吸收的同时才能提升反应性能[57]。TANG等[58]以纯PCN和Mg(NO3)2·6H2O为前驱体通过溶剂热法合成了Mg掺杂的PCN。Mg的存在抑制了电子-空穴的复合,提高了电荷分离效率。在25℃的CO2饱和水溶液中反应6 h后,纯PCN和Mg掺杂PCN产生CH4的量分别为8.17、17.09μmol/g。
4.3 缺陷工程
在材料中引入缺陷是一种常用的调节半导体光催化性能的策略[59]。缺陷的存在会影响材料的光吸收、电荷分离及传递、表面吸附性能等[60]。SHI等[61]采用酒石酸辅助热解双氰胺的方法,制备了含N缺陷的二维片状氮化碳(命名DCN-x,x为酒石酸的质量,单位为g),该N缺陷位于三配位N原子和未缩合的末端氨基上。图5为DCN和DCN-x的能带结构和稳态光致发光光谱(PL)图。由图5看出,缺陷的存在使得带隙减小,增强了材料的光吸收能力,同时促进了电荷的分离。最优的样品(DCN-0.05)在有机液相体系光还原CO2反应5 h后生成CO的量为285μmol/g,较纯DCN的产量(36μmol/g)提升将近8倍。SONG等[62]通过一步热解尿素和聚乙烯亚胺(PEI)混合物,合成了同时具有C和N缺陷的PCN。制备过程中,PEI作为掺杂剂影响了PCN的有序聚合,形成了C缺陷和大量未完全缩合的氨基。体相N缺陷的存在导致了中间能级的形成,提高了光的利用效率,表面C缺陷的存在则促使了末端氨基的形成,碱性的氨基基团有利于材料对CO2的吸附。在水相反应体系、紫外光照条件下,反应4 h,CO和CH4产量分别为32.86、1.68μmol/g,分别是纯PCN的3.2倍和2.5倍。
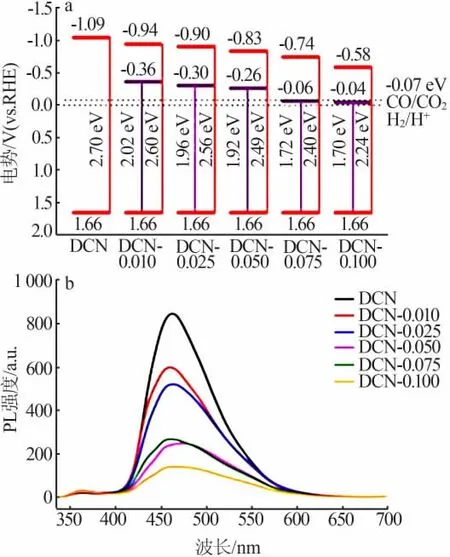
图5 DCN和DCN-x的能带结构(a)和稳态PL图(b)[61]Fig.5 Band structure alignments(a)and steady-state PL spectra for DCN and DCN-x(b)[61]
4.4 构建异质结
半导体异质结是指两种半导体紧密接触的界面。异质结的构建可以改变材料的光吸收性质,促进光生电荷的分离和转移,有利于光催化反应的发生[63]。常见的异质结电荷转移机制分为Ⅱ型和Z型,其中Z型异质结在促进电荷分离的同时,保持了半导体较高的氧化和还原能力,具有更大的优势。ZHAO等[64]通过静电自组装和溶剂热原位硫化法制备了中空In2S3/PCN异质结。由于In2S3和PCN之间形成了紧密的界面,从而电荷分离和传递效率得以提高,如图6所示,电荷传递机制为Ⅱ型。结合中空结构光吸收的优势以及增强的CO2吸附能力,In2S3/PCN光还原CO2性能得到大幅度提升,CO的产生速率为483.4μmol/(g·h),分别是纯IS和PCN的99倍和6倍。JIANG等[65]通过水热法将海胆状α-Fe2O3和PCN两种材料进行复合构建了异质结,海胆状α-Fe2O3的三维分层结构和碱性活性位有利于对CO2的吸附,Z-型异质结的构建有效地促进了电子-空穴对的分离并保留了PCN导带电子的强还原性。在不使用助催化剂和牺牲试剂的情况下,异质结光还原CO2生成CO的速率达到27.2μmol/(g·h),相较于纯PCN提升了2.2倍。SHI等[66]以NH2-MIL-125(Ti)和三聚氰胺为前驱体通过两步热解法制备了富含氧空位的0D/2D TiO2/PCN异质结。在原位热解过程中,半导体间通过化学键形成紧密的相界面。该异质结具有较高的比表面积和孔体积、较高的CO2吸附能力、增强的可见光吸收性能以及较好的电荷分离效率,其光催化还原CO2生成CO产量为77.8μmol/(g·h)。进一步引入过渡金属Co2+修饰异质结制备了Co2+掺杂的TiO2/PCN异质结,Co2+作为界面处连接TiO2和PCN的桥梁,加速了电荷转移,显著提高了电荷分离与传递效率,光催化还原CO2产生CO的速率达290μmol/(g·h),远高于PCN和TiO2/PCN异质结。
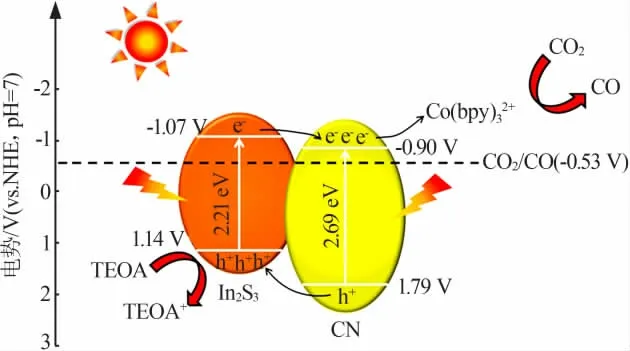
图6 In2S3/PCN异质结光还原CO2机理示意图[64]Fig.6 Proposed CO2 photoreduction mechanism ofIn2S3/PCN heterojunction[64]
5 总结与展望
本文介绍了光催化还原CO2的反应机理以及PCN的几何和电子结构,总结了PCN的制备和改性方法。为了提升PCN光还原CO2的性能,可考虑形貌调控、异原子掺杂、引入缺陷和构建异质结等手段,也可将多种改性策略相结合进一步优化其性能。近年来,PCN在光还原CO2领域得到了广泛的研究和应用,研究者们也提出了多种先进的制备方法和改性手段,使其性能不断提升。
但是仍存在一些问题,例如PCN组成、结构的改变会带来多方面的影响,例如光吸收、电荷分离和CO2吸附能力等,大多数的工作主要是定性地解释性能提升的原因,缺少定量的数据支持。现有的研究主要集中于CO2还原半反应,反应中通常需要加入助催化剂和牺牲剂,体系较为复杂,实现无牺牲剂、无助催化剂条件下将H2O氧化为O2的同时将CO2高效还原为化学燃料的光催化过程仍然是一个挑战。此外,利用熔融盐法、微波法等方法可制备高结晶性的PCN,进而增强光催化活性,因此发展新的制备方法获得高结晶性的PCN也是重要的研究方向。现阶段光催化CO2转化的效率仍然较低,不能满足实际应用的需要,故开发高活性、高选择性的PCN基光催化剂并深入理解反应机理仍然是未来的发展方向。
