野生蒿属植物的DNA分子鉴定
2021-12-13郭国业
郭国业,周 悦
(南阳师范学院 生命科学与农业工程学院,河南省艾草开发利用工程技术研究中心,河南 南阳 473061)
0 引言
蒿属(ArtemisiaLinn.)是菊科(Asteraceae)春黄菊族菊亚族的一个大属,该属植物在亚欧大陆集中分布.我国有187种46变种[1],分布全国.在华北、西北、西南等地多生长在草场、荒漠、森林、戈壁以及高山草场,在华东、华中、华南地区的蒿属植物多生长于荒野、道路旁以及海滩与丛林.蒿属植物的开花期在8~9月份,常有浓郁的挥发性香气;多为草本,少数为灌木、半灌木.有许多种类是重要或常用的药草植物,可以当作利尿剂、化痰剂、抗炎症药、止血剂、低血压药、抗过敏药或艾灸用药,少数种供食用.有的植物根系粗大,耐盐碱,茎和枝耐沙,可以作为抗御风沙的先锋植物或者辅助植物[2].河南蒿属植物资源较为丰富,常见的有艾(Artemisiaargyi)、黄花蒿(Artemisiaannua)、青蒿(Artemisiacarvifolia)、茵陈蒿(Artemisiacapillaris)、秦岭蒿(Artemisiaqinlingensis)、蒙古蒿(Artenmisiamongolica)、南毛蒿(Artemisiachingii)等,多分布于山地、河坡、湿地、树下、荒野及城区绿化地带.
DNA条形码(DNA barcoding)于2003年首次提出[3],多用于物种的快速识别鉴定和系统进化研究等方面.目前,DNA条形码技术在生命科学领域已经成熟并得以广泛应用,是物种谱系地理学研究和资源识别的重要工具[4].与传统的形态分类学相比,DNA条形码具有更加高效、准确的物种识别效率[5],特别是在中药资源鉴定、药材流通体系等方面应用广泛[6-9].近年来,DNA条形码序列分析广泛应用于植物的分类鉴定和系统亲缘关系的分析,成为探索种质关系的有效手段.目前,菊科中的已有多个属种植物利用DNA条形码基因来探究属内种间关系,但在蒿属植物中的应用研究相对较少.蒿属植物的系统分类学研究主要集中在形态分类上,在分子水平上的研究很少,急需通过分子鉴定以明确蒿属植物的分类关系.
本研究利用DNA条形码基因,对在南阳市白河湿地及其周边区域分布的9个野生蒿属植物的基因组进行序列变异分析、遗传距离计算、构建NJ系统进化树,旨在探究和分析蒿属植物的种间亲缘关系,以期为艾草植物的遗传育种和开发利用提供参考依据.
1 材料与方法
1.1 实验材料
实验材料为2020年10月,于河南南阳市区白河湿地公园及其周边区域所采集的野生蒿属植物的新鲜幼嫩叶片,并制成简易标本保存.植物标本依据《中国植物志》进行形态学特征识别,并由中国科学院植物研究所高天刚研究员和南阳师范学院生命科学与农业工程学院李景照教授识别鉴定,材料确定为黄花蒿(Artemisiaannua)、南毛蒿(Artemisiachingii)、野艾蒿(Artemisialavandulifolia)、蒙古蒿(Artemisiamongolica)、艾(Artemisiaargyi),所有试验材料均为野生种(表1).

表1 蒿属植物种质资源信息
1.2 植物基因组DNA的提取
对9个蒿属植物样品进行植物基因组DNA提取.分别称取200 mg新鲜采集的植物叶片组织,液氮研磨.利用植物基因组DNA提取试剂盒进行基因组DNA提取[10].使用1.0 %的琼脂糖凝胶电泳和凝胶成像系统(美国-BIO)检测和成像.对样品基因组检测质量不佳或显示无带的样本重新提取,将提取的蒿属植物基因组DNA 保存于-20 ℃.
1.3 引物筛选
实验所用引物由华大基因公司(BGI)合成.利用变异速率较高且通用性强的19对候选 DNA 条形码基因对蒿属植物DNA进行PCR预扩增和筛选[11].以Tranks 2K分子 Maker为对照,利用候选条形码基因对部分蒿属植物的样品材料DNA进行PCR扩增.将PCR扩增产物进行凝胶电泳检测和成像,成功获得目的基因的单一克隆.最终筛选出 ITS2和psbA-trnH作为本实验蒿属植物的条形码鉴定基因(表2).

表2 ITS2、psbA-trnH引物序列
1.4 PCR扩增、检测及测序
通过对PCR反应体系和条件的优化,确定最适反应体系及扩增程序,分别见表3和表4.基于所确定的最适PCR反应体系和扩增程序,利用ITS2和psbA-trnH基因对所采集的9份蒿属植物样本进行PCR扩增,扩增产物通过凝胶电泳检测,利用凝胶成像系统成像,对单一克隆产物测序.

表3 PCR反应的最佳体系

表4 PCR反应程序
1.5 数据分析
将测序得到的条形码基因序列在NCBI基因数据库中进行Blast以验证获取序列的正确和准确度.利用MEGA基因分析软件分别对trnH序列、ITS序列以及联合基因序列进行变异位点、简约信息位点、核苷酸置换率和遗传距离等的分析,采用 NJ 邻接法(Neighbor-Joining,NJ)构建系统进化树[12-13].
2 结果分析
2.1 DNA提取及PCR扩增
利用植物基因组试剂盒成功提取出9种蒿属植物叶片的基因组DNA.基于ITS2和psbA-trnH基因引物均成功扩增出所有的植物材料的DNA片段,扩增产物大小为450 bp左右的单一明亮条带,且扩增和测序的成功率均为100%.图1为基于psbA-trnH和ITS2条形码基因的蒿属植物PCR 扩增凝胶电泳检测结果.
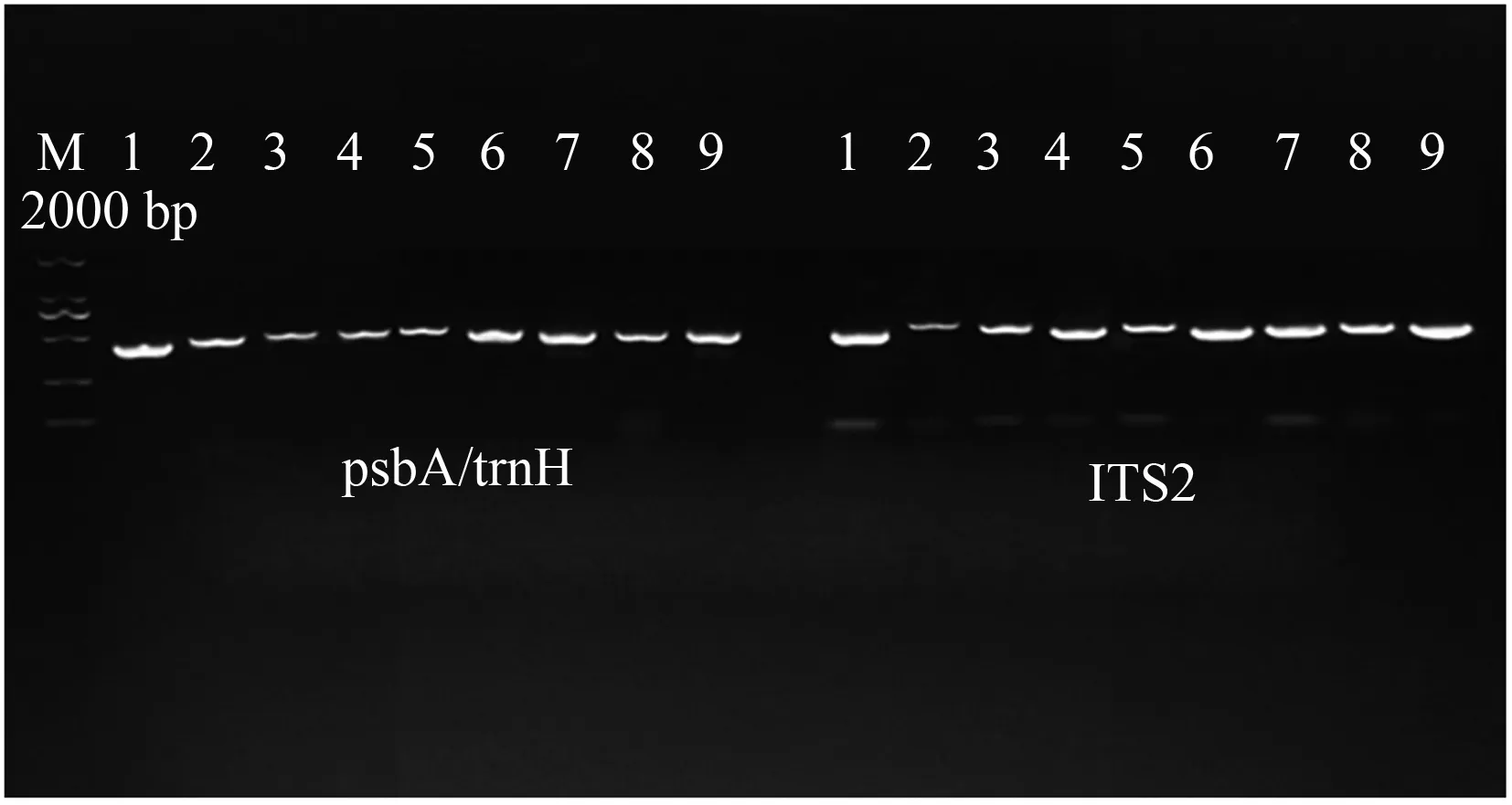
图1 基于psbA-trnH和ITS2条形码基因的蒿属植物PCR扩增检测图
2.2 ITS2和psbA-trnH基因序列变异分析
将测得的9条ITS2序列和psbA-trnH序列进行校正和比对分析,剪切两端不可信序列,最终获得ITS2序列总长为445 bp,psbA-trnH基因序列大小为441 bp.基因变异分析结果显示,ITS2基因序列具有419个保守位点,19个变异位点,11个分离位点;psbA-trnH基因序列具有435个保守位点,6个变异位点,6个分离位点.
2.3 基于ITS2+psbA-trnH联合基因的序列变异分析
基于psbA-trnH+ ITS2联合基因的序列变异分析结果显示(表5).联合基因共有886个位点,缺失位点有19个,共有保守位点845个,多态位点22个,可变位点15个,简约信息位点7个.联合基因核苷酸多态性π为 0.008,显示ITS2和psbA-trnH的联合基因在蒿属植物资源中具有较高的进化速率;基于ITS2与psbA-trnH的联合基因的Tajima’D中性检验为-0.702,显示种群基因变异偏离中性突变,在群体进化中可能遭遇了选择压力.分析数据显示,将ITS2条形码基因与psbA-trnH基因进行联合数据分析表现出较高的序列核苷酸多样性.

表5 psbA-trnH+ITS2联合基因序列变异
2.4 基于ITS2+psbA-trnH联合基因的系统进化分析
基于ITS2和psbA-trnH两个基因的联合分析具有更高的变异,适于系统进化树重建.图2为基于ITS2基因和psbA-trnH两个基因的联合并采用NJ邻接算法重建蒿属植物的系统进化树.结果显示,9个蒿属植物重建的系统进化树形成三个明显的进化分支(图2).分支Ⅰ包含艾、南毛蒿、野艾蒿和蒙古蒿,其中艾与野艾蒿同属一个分支,野艾蒿与南毛蒿同属一个分支,蒙古蒿与野艾蒿聚类,表明艾、野艾蒿和蒙古蒿植物具有较近的进化亲缘关系.分支Ⅱ为1个南毛蒿植物单独聚类,南毛蒿属于蒿属植物腺毛蒿组的多花蒿系,显示该组植物与艾组植物的进化关系较远,进化过程较为独立.分支Ⅲ为1个艾蒿组(Sect.Abrotanum)黄花蒿系的黄花蒿植物,显示该组植物与艾组植物在进化关系上存在明显的遗传分歧.

图2 基于 ITS2+psbA-trnH联合基因的蒿属植物系统进化树注:分支前的数字为自然支持值,分支后的数字为实验材料编号
3 讨论
DNA条形码技术在植物学系统进化研究中起着重要作用.它以 DNA 序列为检测对象,样本选择不受时节和环境的限制,有效扩大了样本的选择范围[14].本实验结果表明,不同的蒿属植物具有较丰富的遗传多样性.依据《中国植物志》的系统地位划分,艾、野艾蒿、蒙古蒿都属于蒿属植物中的艾组.其中,艾属于艾组中的真艾系,蒙古蒿属于艾组的艾系,野艾蒿属于艾组的野艾蒿系,三种都同属艾组,在系统进化树上能够准确聚为一个大分支,显示三者亲缘关系较近,与形态学分类地位一致.南毛蒿属于蒿属植物腺毛蒿组的多花蒿系,在《中国植物志》第76(2)卷中记录了在河南南部有南毛蒿植物的分布,但在《河南植物志》(第3册)里并未给予明确报道,李春凤等通过调查采集及整理鉴定河南菊科蒿属植物标本,对《河南植物志》蒿属植物的南毛蒿进行了增补修订和形态介绍[15-17].在本研究中,系统进化树的分支Ⅰ中一个南毛蒿植物与艾组植物聚类,而5号南毛蒿植物则形成单独聚类分支Ⅱ,显示南毛蒿植物所在的腺毛蒿组植物与艾组植物存在一定的遗传距离,后续将结合微形态学特征鉴定、细胞染色体分析和分子系统学方法,综合分析基于分子进化树所产生的两组蒿属植物的系统关系是否能够表明在进化过程中发生了种间杂交和基因交流.依据中国植物志的系统分类地位,黄花蒿植物属于艾蒿组的黄花蒿系,本研究的系统进化树中显示,黄花蒿植物单独聚类在最外分支,显示与其他蒿属植物间存在明显的遗传分歧,进一步验证了黄花蒿与其他艾组植物如艾、野艾蒿和蒙古蒿的亲缘关系较远.
本研究基于ITS2、psbA-trnH条形码基因的联合数据分析可以有效地实现对蒿属植物进化亲缘关系的分析.目前,蒿属植物部分物种的分类地位和关系依旧存在争议,本研究结论可以为鉴定蒿属植物的种间亲缘关系和分类提供一定的参考依据,对于开发蒿属植物鉴定的DNA条形码基因具有一定的参考意义.
