基于RNA-Seq技术分析不同生物被膜形成能力的临床耐药鲍曼不动杆菌基因表达差异*
2021-12-13刘巍巍国果吴兆颖毛成菊李忠旬贾利娜尚小丽彭建吴建伟
刘巍巍,国果,吴兆颖,毛成菊,李忠旬,贾利娜,尚小丽,彭建**,吴建伟**
(1.贵州医科大学 基础医学院 人体寄生虫学教研室,贵州 贵阳 550025;2.贵州医科大学 基础医学院 现代病原生物学特色重点实验室,贵州 贵阳 550025;3.贵州医科大学 生物工程学院,贵州 贵阳 550025)
鲍曼不动杆菌(Acinetobacterbaumannii,AB)生物被膜是疾病发病机理中重要的毒力因子[1]。与生物被膜感染相关的疾病发病率不断上升,且治疗更加困难。最常见的生物被膜感染包括口腔牙周炎、慢性中耳炎、鼻窦炎、尿路感染以及囊性纤维化患者的肺部感染等[2]。细菌通过附着在宿主组织(牙齿、黏膜表面等)形成生物被膜,抗生素难以彻底清除,导致慢性反复感染[3];另一种情况是细菌对非生物表面的粘附,如在留置医疗器械或生物材料(中央静脉导管、气管导管、人工心脏瓣膜、隐形眼镜等)表面形成生物被膜,并且细菌会从生物被膜上脱落并扩散到机体周围组织或血液中,进一步加剧感染[4]。研究表明,生物被膜表型是异质性的,不同的菌株能形成不同的生物被膜,白色念珠菌临床分离株形成生物被膜强和弱的菌株导致的感染预后不同,与死亡率显著相关[5]。研究报道,白色念珠菌中高生物被膜形成能力的菌株比低生物被膜形成能力的菌株具有更高的致病性[6]。与环境菌株相比,鲍曼不动杆菌临床分离菌株具有更高的形成强生物被膜的能力[7]。对100株分离自重症监护病房的鲍曼不动杆菌进行检测,结果发现58%的菌株表现出很强的生物被膜形成能力,且所有强生物被膜形成能力的菌株都为泛耐药菌株[8]。临床耐碳青霉烯类鲍曼不动杆菌(carbapenem-resistant AB,CRAB)是一种类似耐甲氧西林金黄色葡萄球菌的超级耐药菌,其形成生物被膜后对抗生素的耐药性进一步提高[9]。本研究中,采集临床分离CRAB,通过对生物被膜形成能力强和弱的菌株进行转录组测序,比较两种不同生物被膜形成能力菌株的基因表达差异,探究导致其生物被膜形成差异的原因,以期为临床CRAB生物被膜形成机制以及感染的防治提供依据。
1 材料与方法
1.1 材料
1.1.1菌株 CRAB 4、13、55、78、117、177、191和196号菌株分离于临床住院患者痰液、血液样本。大肠埃希菌ATCC25922、铜绿假单胞菌ATCC27853为质控菌,由贵州医科大学病原生物学实验室保存。
1.1.2实验试剂与仪器 Mueller-Hinton(MH)肉汤培养基、Trypticase Soy Broth(TSB)肉汤培养基和Luria-bertani(LB)固体培养基、结晶紫粉末均购自北京索莱宝公司,溶菌酶(购自上海生工公司)、SYBR Premix Ex TaqTMⅡ(大连宝生物公司),RIZOL(美国invitrogen公司)。恒温培养箱购自上海博讯实业,细菌比浊仪购自上海昕瑞有限公司,高压蒸汽锅购自日本ALR公司,微量加样枪、PCR扩增仪、速冷冻离心机购自德国Eppendorf公司,ABI7300荧光定量PCR仪购自美国ABI公司,NanoDrop ND-2000分光光度计(美国Thermo公司)。
1.2 方法
1.2.1临床菌株鉴定及生物被膜的培养和收集 细菌的鉴定与药敏试验均采用法国生物梅里埃VITEK-2全自动微生物分析仪进行分析,同时对菌株的16SrRNA和rpoB基因进行扩增,并对PCR产物进行测序和分析,以保证菌株鉴定的准确性。选取生物被膜形成能力强(CRAB 4、55、78、117)的菌株作为强生物被膜(biofilm,BF)组,作为4个生物学重复,菌株在形态和特性上相似。取培养至对数期的菌液,用TSB培养基稀释至每毫升106cfu。取菌液4 mL加入6孔培养板中,置于37 ℃恒温箱中培养24 h,然后弃去游离菌液,PBS清洗后用细胞刮刀刮取生物被膜菌。选取生物被膜形成能力弱(CRAB 13、177、191、196)的菌株作为弱生物被膜(weak biofilm,WBF)组,作为4个生物学重复,菌株在形态和特性上相似,余按BF组方法培养并收集生物被膜菌。
1.2.2RNA提取与质量检测 参考文献[10]的方法,先使用溶菌酶对样品进行预处理,再加入TRIZOL溶液裂解,冰上静置15 min。加入氯仿200 μL混匀,冰上静置5 min,4 ℃,12 000 r/min离心15 min。RNA位于上层水相中,小心吸取上清液,转入新的离心管中。加入等体积的异丙醇混匀,室温静置10 min,4 ℃,12 000 r/min离心10 min,弃上清,加入75%乙醇(预冷)1 mL,轻轻吹打混匀,溶解沉淀,4 ℃,7 500 r/min离心5 min,弃上清,室温干燥10 min。加入焦炭酸二乙酯(diethyl pyrocarbonate,DEPC)水30~50 μL溶解沉淀,分装,置于-80 ℃保存。琼脂糖凝胶电泳检测RNA降解情况,通过分光光度计(NanoDrop ND-2000)测定RNA浓度。
1.2.3文库构建和转录组测序(RNA-Seq) 提取到的RNA经检测合格后,去除Total RNA中的核糖体RNA(rRNA),获得mRNA。随后加入fragmentation buffer将得到的mRNA随机打断成短片段,按照链特异性建库的方式建库[11]。文库构建完成后,先使用Qubit2.0 Fluorometer进行初步定量,随后使用Agilent 2100 bioanalyzer对文库的insert size进行检测,insert size符合预期后通过实时荧光定量PCR(quantitative Real-time PCR,qRT-PCR)对文库有效浓度进行准确定量,以保证文库质量。库检合格后,把不同文库按照有效浓度及目标下机数据量的需求pooling后进行Illumina测序。
1.2.4数据分析 以鲍曼不动杆菌AB030(NZ_CP009257.1)的基因组作为参考基因组,用Bowtie2软件进行基因组定位分析[12]。HTSeq v0.6.1用于计算映射到每个基因的读数,使用DESeq软件分析两个比较组合之间的差异表达(每个组4个生物学重复)。通过DESeq统计程序,将基于负二项式分布的模型确定数字基因表达数据中的差异表达。使用Benjamini和Hochberg的方法来调整所得P值,设置差异基因筛选的标准[13]:校正后的P值 padj<0.05,且|log2(fold change)|>1。对差异表达基因(differentially expressed genes,DEGs)进行基因本体(geneontology,GO)功能注释和京都基因与基因组百科全书(kyoto encyclopedia of genes and genomes,KEGG)通路富集分析。
1.2.5实时荧光定量PCR验证差异基因 从转录组测序鉴定得到的DEGs中挑取8个差异基因(表1),利用qRT-PCR验证差异基因。按之前所述方法提取RNA后,用逆转录试剂盒将提取的总RNA逆转录成cDNA。PCR扩增总反应体系20 μL(cDNA模板2.0 μL、SYBR®Green Realtime PCR Master Mix 10 μL、ROX Reference Dye(50×)0.4 μL、ddH2O 6.0 μL、上下游引物各0.8 μL)。反应条件:95 ℃预变性30 s,95 ℃变性5 s,60 ℃退火31 s,共40个循环。反应在ABI7300荧光定量PCR仪上进行,以AB 16SrRNA为内参基因,目的基因的相对表达量采用2-ΔΔCT方法计算。

表1 Real-time PCR引物Tab.1 Real-time PCR primers
2 结果
2.1 RNA质量
总RNA条带完整清晰,无杂带,样本无污染(图1)。通过Agilent 2100 bioanalyzer精确检测显示,RIN值8~10,RNA完整性好;检测RNA纯度,OD260/280为1.8~2.2,OD260/230>1.8。样品满足后续建库和试验要求。见表2。
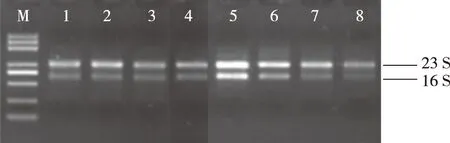
注:M为marker条带,1~4为BF组总RNA,5~8为WBF组总RNA。图1 RNA琼脂糖凝胶电泳结果Fig.1 RNA sepharose gel electrophoretogram
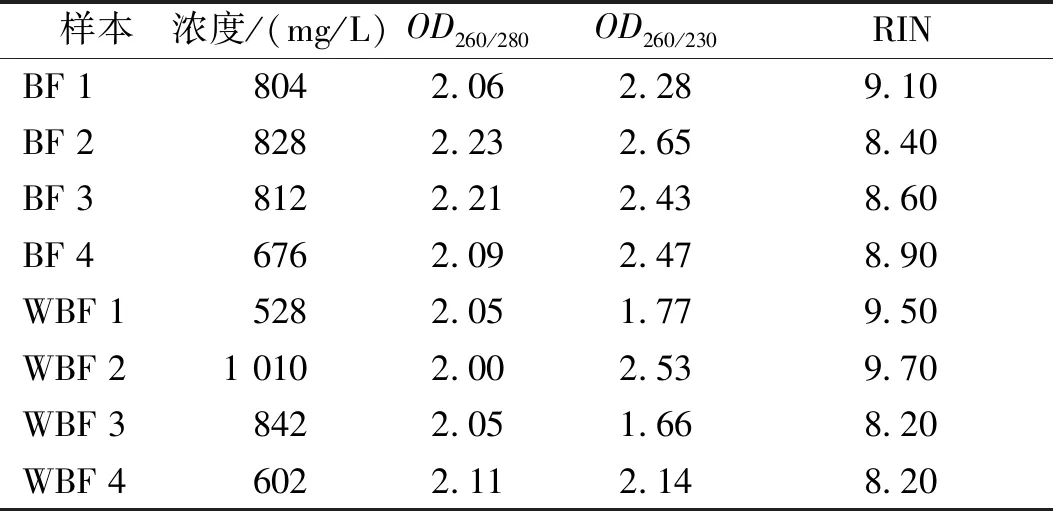
表2 RNA样品质量信息Tab.2 RNA sample general information
2.2 mRNA测序数据
样品测序结果显示错误率为 0.03%,显示测序结果可靠。每组样品的 clean reads 数据>1G ,测序深度足够覆盖所有可能存在的基因。Q20>97%,Q30>92%,实验所产生的测序reads成功比对到基因组的比例高于82%,其中具有多个定位的测序序列占总体的百分比<7%,符合后续分析的要求。见表3。
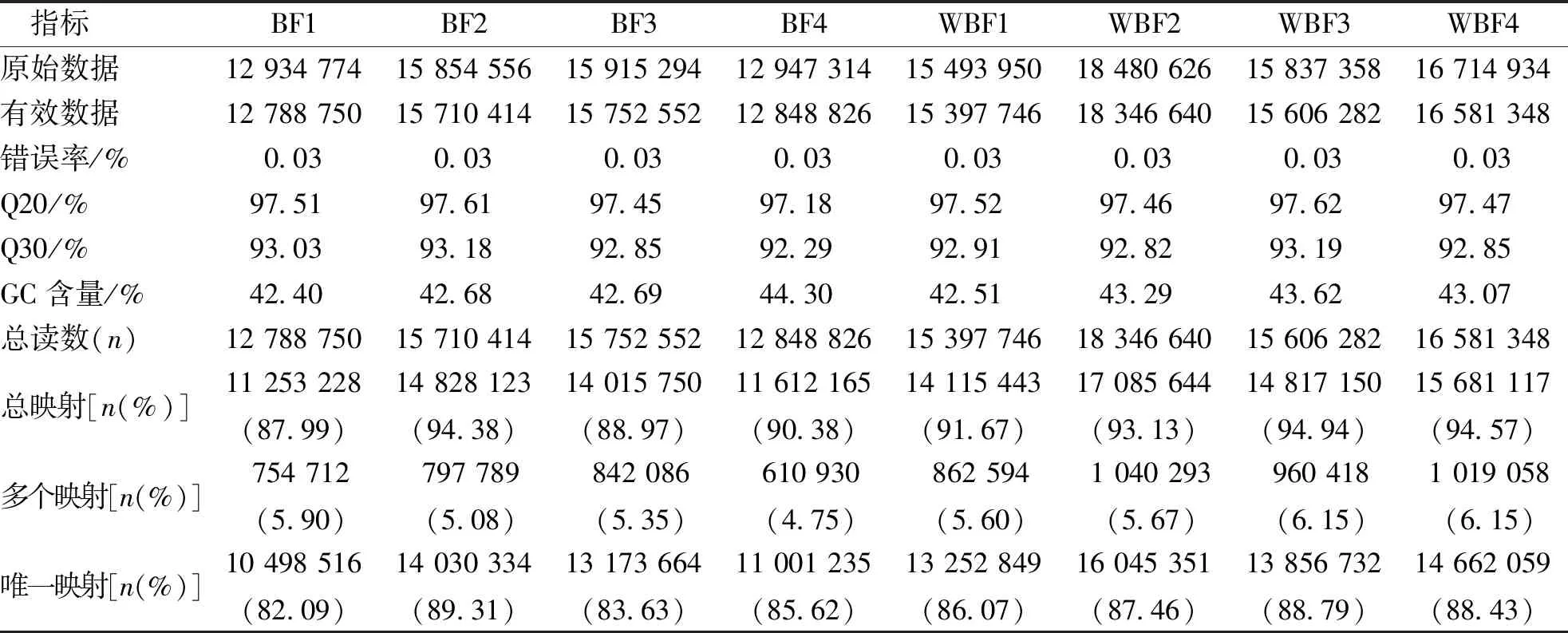
表3 各样品mRNA测序数据Tab.3 Sample mRNA sequencing data
2.3 基因表达差异分析
DEGs的火山图显示,与WBF组比较,BF组有171个基因差异表达,包括106个上调(红色)的基因和65个下调(绿色)的基因(图2)。ATP相关基因:LamB/YcsF家族蛋白(IX87_RS04005)参与ATP水解等过程,其 log2(fold change)值为-3.48;ATP合酶亚基α(IX87_RS15035)、ATP合酶亚基β(IX87_RS15045)、ATP合酶亚基γ(IX87_RS15040)、ATP合酶亚基δ(IX87_RS15030)的log2(fold change)值分别为-2.1、-2、-2.3和-2.2。DNA结合转录调节相关基因:DNA结合蛋白(IX87_RS19850)的log2(fold change)值为3.8;AraC家族转录调节因子(IX87_RS19260)、XRE家族转录调节子(IX87_RS02480)、Lrp/AsnC家族转录调节因子(IX87_RS02640)均为DNA结合转录调节相关基因,log2(fold change)值分别为2.7、2.1和1.7。菌毛合成相关基因:菌毛合成调控相关蛋白CsuA(IX87_RS06910)的log2(fold change)值为2.6。膜相关基因:外膜蛋白(IX87_RS01665)的log2(fold change)值为-2.1;外膜孔蛋白OmpW家族蛋白(IX87_RS06185)参与铁的吸收和黏附素的结合过程,log2(fold change)值为2.4。上调的DEGs主要涉及DNA结合、膜蛋白、菌毛调控相关基因等,下调的DEGs主要涉及ATP的合成和分解、外膜蛋白等。见图2。
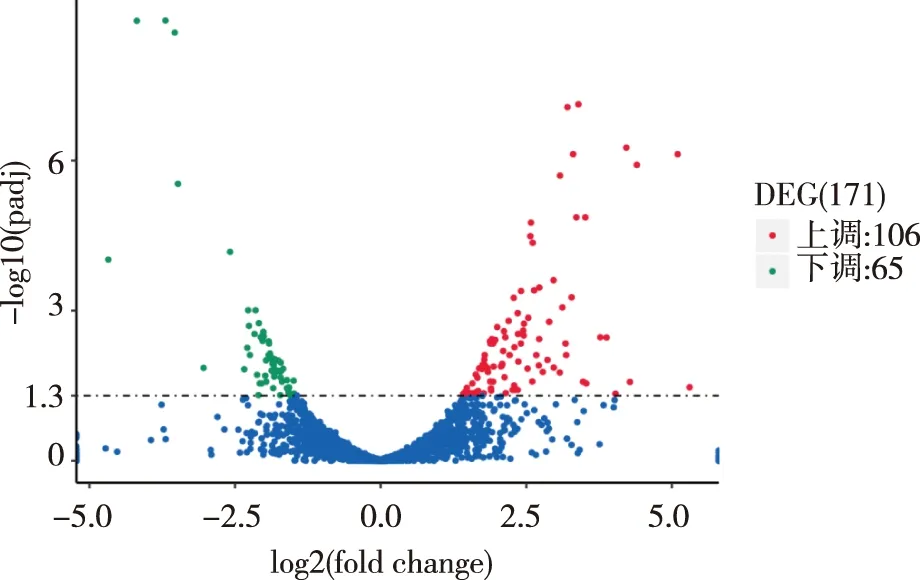
注:红、绿点分别为上调和下调的DEGs,蓝点为不显著DEGs。图2 DEGs火山图Fig.2 DEGs volcanoplot
2.4 差异基因GO富集分析
GO富集分析结果显示最显著的30个GO条,如阳离子跨膜转运(cation transmembrane transport)、质子跨膜转运(proton transmembrane transport)、嘌呤核糖核苷酸代谢过程(purine ribonucleotide metabolic process)、嘌呤核苷酸的生物合成过程(purine nucleotide biosynthetic process)、核糖核苷酸代谢过程(ribonucleotide metabolic process)、核糖核苷酸的生物合成过程(ribonucleotide biosynthetic process)、ATP代谢过程(ATP metabolic process)及ATP生物合成过程(ATP biosynthetic process)等显著富集于生物过程;质子转运双扇形ATP酶复合物,催化结构域(proton-transporting two-sector ATPase complex,catalytic domain)及质子转运ATP酶复合物(proton-transporting ATP synthase complex)显著富集于细胞组分;ATP酶活性,与离子的跨膜转运耦合、旋转机制(ATPase activity,coupled to transmembrane movement of ions,rotational mechanism)、活性离子跨膜转运蛋白活性(active ion transmembrane transporter activity)等显著富集于分子功能类别。见图3。
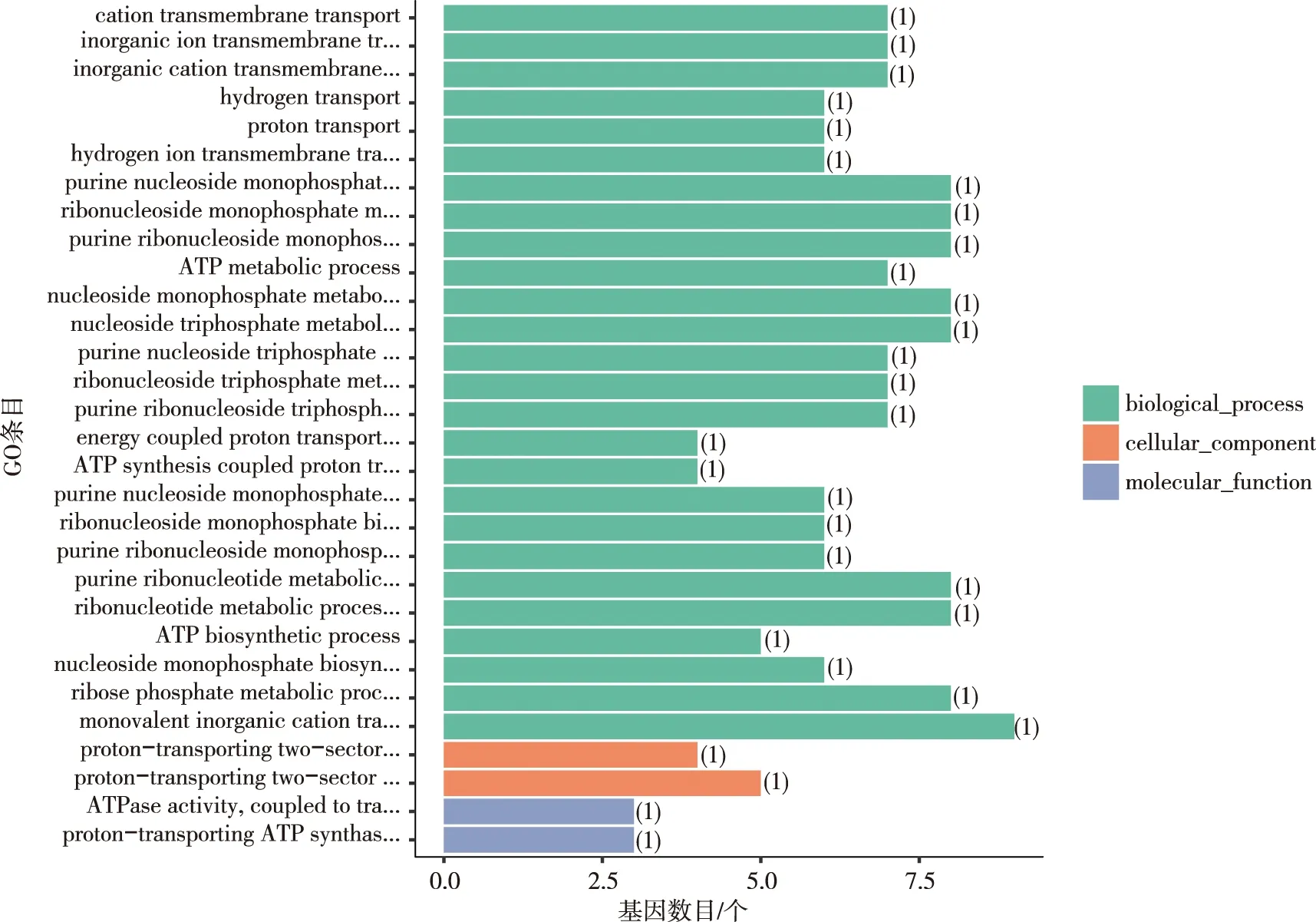
注:(1)P<0.05。图3 DEGs的GO富集分析Fig.3 GO enrichment analysis of DEGs
2.5 差异基因KEGG富集分析
KEGG富集分析结果表明,DEGs参与的KEGG代谢通路中最显著的通路有20条,分别为缬氨酸、亮氨酸和异亮氨酸的降解(valine,leucine and isoleucine degradation)、氧化磷酸化(oxidative phosphorylation)、糖酵解/糖异生(glycolysis/gluconeogenesis)、β-丙氨酸代谢(beta-alanine metabolism)、泛酸和CoA生物合成(pantothenate and CoA biosynthesis)、代谢途径(metabolic pathways)、香叶醇的降解(geraniol degradation)、RNA降解(RNA degradation)、丙酮酸代谢(pyruvate metabolism)、丙氨酸、天冬氨酸和谷氨酸代谢(alanine,aspartate and glutamate metabolism)、组氨酸代谢(histidine metabolism)、精氨酸和脯氨酸代谢(arginine and proline metabolism)、半乳糖代谢(galactose metabolism)、柠檬烯和蒎烯降解(limonene and pinene degradation)、硫代谢(sulfur metabolism)、抗生素的生物合成(biosynthesis of antibiotics)、脂肪酸代谢(fatty acid metabolism)、万古霉素耐药(vancomycin resistance)、肽聚糖的生物合成(peptidoglycan biosynthesis)和甲烷代谢(methane metabolism)。见图4。
2.6 DEGs的验证
从转录组差异基因中挑选8个基因,其中上调和下调表达的基因分别为5和3个;采用qRT-PCR验证不同生物被膜形成能力的两组菌株的差异,该结果与转录组结果趋势一致,表明转录组数据可靠。见图5。
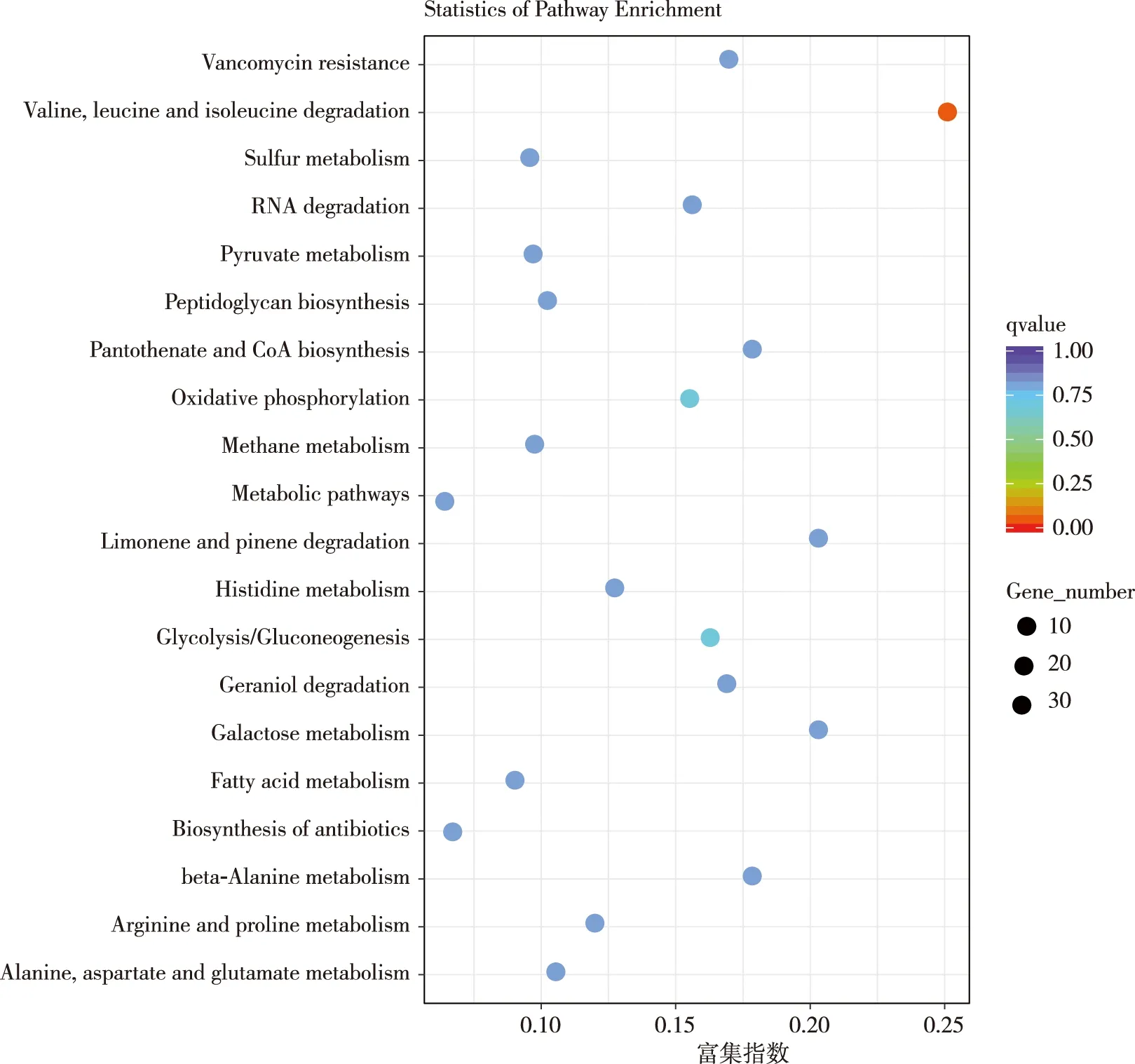
注:横坐标为基因富集指数,纵坐标为信号通路名称。图4 DEGs的KEGG富集分析Fig.4 KEGG enrichment scatter plot of DEGs
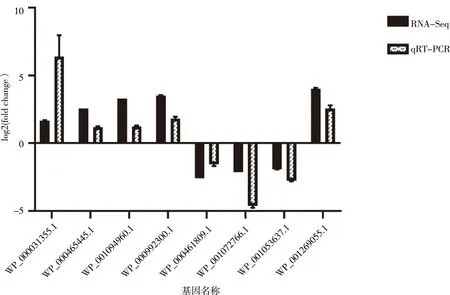
图5 8个差异基因表达水平的比较(RNA-Seq、qTR-PCR)Fig.5 Expression of 8 differential genes(RNA-Seq,qTR-PCR)
3 讨论
细菌耐药问题一直备受关注,研究表明,细菌在受到抗菌药物等外界压力刺激下会发生耐药基因的变化,上调耐药基因的表达或进化以获得某些特性,导致多重耐药、泛耐药菌株的克隆流行[14-15]。近年来,CRAB的检出率逐年上升,已被多次报道[16-17]。鲍曼不动杆菌形成生物被膜是其重要的毒力因子,与一般的细菌导致的感染相比,生物被膜能为菌体提供持续的防护,帮助细菌适应不利的外环境和躲避宿主的攻击,形成稳定释放细菌的慢性感染源,导致感染反复发生,同时会增强对抗菌药物的抵抗能力,可由敏感变成耐药,给治疗带来更大的阻碍[18-19]。了解临床CRAB形成不同生物被膜的分子机制,对其导致的严重感染的防治和诊断等研究有着重要的意义。
转录组测序是研究基因表达变化的重要手段,转录组研究能够从整体水平研究基因功能以及基因结构,揭示特定的生物学过程,已广泛应用于基础研究、临床诊断和药物研发等领域[20]。经RNA-Seq分析,将生物被膜形成能力强和弱的临床分离CRAB生物被膜态菌进行比较,获得171个DEGs,其中106个基因为上调表达,65个基因下调表达;随机挑取了8个差异基因进行qRT-PCR验证,其结果与转录组测序结果一致,表明测序结果可靠。DEGs主要包括ATP水解和合成相关基因、DNA结合转录调控因子、外膜蛋白以及菌毛调控基因。值得注意的是,与生物被膜形成能力弱组相比,生物被膜形成能力强组中菌毛调控相关基因CsuA表达上调,表明菌毛加快合成,促进生物被膜的形成。细菌对宿主表面的粘附是形成生物被膜的第一步。菌毛则介导了细菌对非生物表面的粘附,帮助细菌在新的表面定殖[21]。研究表明,外膜蛋白参与生物被膜的形成,特别是在初始粘附阶段发挥作用;相对于非生物表面,外膜蛋白OmpA被证明对于鲍曼不动杆菌19606粘附在生物表面的能力至关重要[22-23]。并且,CarO和OmpA等膜蛋白能和碳青霉烯酶OXA-23发生相互作用,从而提高对抗生素的耐药性[24]。对多重耐药鲍曼不动杆菌菌株与敏感菌株进行蛋白质组学比较,结果发现孔蛋白OmpW在多重耐药菌株BAA-1605中显著上调[25]。本研究中,与WBF组相比,BF组外膜蛋白基因(IX87_RS01665)表达下调,孔蛋白OmpW基因(IX87_RS06185)表达上调。这表明膜蛋白相关基因的变化影响临床CRAB形成不同的生物被膜,且影响其耐药性。GO富集分析显示差异表达基因显著富集到阳离子跨膜转运、质子跨膜转运、核酸及ATP的合成和代谢等生物合成过程中。KEGG富集分析主要涉及细菌的生化代谢途径的改变,如各种氨基酸的代谢合成、氧化磷酸化等。以上结果都表明菌株之间的生化代谢途径的改变对生物被膜形成能力的强弱有影响,而抗生素的生物合成、万古霉素耐药通路的变化表明细菌生物被膜形成能力与其对抗生素的耐药性有关。
本研究通过转录组技术,探讨临床CRAB生物被膜形成能力不同的菌株之间基因表达的差异,鉴定出信号传导和代谢过程等途径相关基因,下一步,将选择部分基因进行验证和功能分析,从而为后续的分子机制研究提供一定的理论基础。
