甘油磷酰胆碱的分析、制备及纯化研究进展
2021-11-30邹俊康鲍宗必杨启炜张治国任其龙杨亦文
邹俊康,鲍宗必,杨启炜,张治国,任其龙,杨亦文
(1 浙江大学化学工程与生物工程学院,生物质化工教育部重点实验室,浙江 杭州 310027;2 浙江大学衢州研究院,浙江 衢州 324000)
甘油磷酰胆碱的全名为L-α-甘油磷酰胆碱(L-α-glycerylphosphorylcholine,GPC),是人体内卵磷脂分子上的两条脂肪酸链水解后的产物[1]。早在1945 年Schmidt 等[2]便从牛的胰脏中提取出了GPC 粗品,第一次确认了GPC 的结构。由于其结构特性,GPC有助于细胞膜磷脂的合成,从而增强细胞膜的流动性,同时还会对脂质代谢产生积极影响[3]。
GPC能够穿过血脑屏障,为神经递质乙酰胆碱的合成提供原料,从而提高海马体中乙酰胆碱的水平,有效地改善人体胆碱状态[4],并且通过增加神经细胞的形成、减少神经元的死亡来支持大脑和神经系统的功能[5-6],提升大脑认知能力[7]。此外,GPC能提高基因的表达水平,预防由衰老造成的认知功能下降[8],这也使得GPC 在治疗阿尔兹海默症[9-10]、健忘症[11]、脑缺血型中风[12]等精神性疾病上有明显的效果。Strifler等[13]证实GPC是一种靶向线粒体的化合物,能够减少啮齿动物体内自由基含量,减轻缺血再灌注引起的损伤和炎症反应[3,14],具有改善心血管疾病的作用[15]。Szabó等[16]发现GPC能降低辐射诱导的形态学损伤和致死性,显示出防辐射的作用。Izu 等[17]发现GPC 与S-腺苷蛋氨酸同时使用能够降低糖尿病模型小鼠的血糖,显示出治愈糖尿病的可能性。
GPC被认为是无毒且安全的化合物[18],不仅具备治愈各种疾病的潜力,还被广泛应用于食品、保健品和化妆品等行业,展现出良好的市场前景[19]。但是,国内高纯度药用GPC 几乎全部依赖进口。为进一步促进GPC 相关产品的研究,本文综述了最近几年来GPC 的研究现状,重点介绍了GPC 的分析、制备和纯化技术的研究进展。
1 GPC的分析方法
早在1990 年,意大利就有GPC 上市,波兰、阿根廷、韩国、俄罗斯等国家也陆续上市了该药品。但在国内,GPC作为一种三类新药,还没有上市。在研究GPC 及其相关产品时,建立全面的分析检测方法至关重要。赵艳艳[20]采用核磁共振波谱(NMR)、质谱(MS)、红外光谱(IR)、紫外光谱(UV)等光谱学技术系统地测定了GPC的光谱学数据,确定了GPC 的结构及化学位移归属(图1)。这些传统方法能定性分析GPC 的存在与否,但无法准确检测出GPC 的含量。分析检测得到GPC 浓度和纯度对于探究其产品的质量更为重要,因此,下面将从薄层色谱法、核磁共振谱法、滴定法和液相色谱法来综述GPC的定量分析方法。
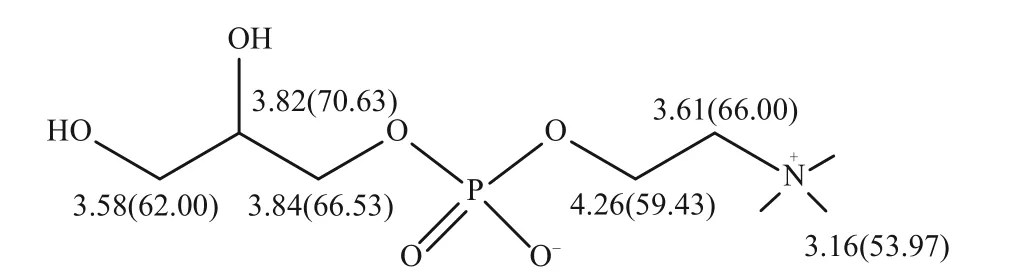
图1 GPC的化学结构及其化学位移归属[20]
1.1 薄层色谱法
薄层色谱由于其操作简便、快速等特点,通常被用作一种定性检测方法,用于判断待测物是否存在。为测定磷酰胆碱酯交换反应所制备的GPC 含量,杜旭佳[21]用硅胶粉和甲基纤维素钠混合物作为固定相,用5mol/L 的氨水和正丙醇的混合液作为展开剂,按照正丙醇、氨水体积比13∶7配制。经过点样、展开和染色后使用Adobe Photoshop 软件对斑点的表面积进行计算,以GPC浓度为横坐标,斑点面积为纵坐标,建立标准曲线,从而测定GPC的浓度。但此方法对斑点表面积的计算误差大,使所测得GPC 的浓度不准确,可作为中控方法,判断反应过程中是否有GPC的生成。
1.2 核磁共振谱法
核磁共振谱法是对物质的结构进行定性分析的强有力工具,Billadello等[22]利用核磁共振磷谱定量分析了组织中的GPC 含量,此方法可以有效测定组织中GPC 的含量,且回收率较高,但此方法检测结果的变异系数太大,导致所测得含量误差大。为准确分析大豆卵磷脂水解产物中各杂质的含量,杜章斌等[23]利用核磁共振氢谱法准确定位了GPC、卵磷脂(PC)和溶血磷脂(LPC)的特征质子峰,并根据每个特征性质子峰的相对面积与物质的量的对应关系计算出三种物质的相对含量,此方法可有效避免样品中水分对检测结果的干扰,使测定结果更准确。但核磁共振氢谱的图谱较为复杂,并且测量特征性质子峰的峰面积存在误差,使得所测量的GPC含量不够准确。为提高测量的准确率,黎玲玲等[24]成功建立核磁共振磷谱定量法,以磷酸三苯酯为内标,以MEOD-d4作溶剂,以(样品/内标)的质量比为横坐标,NMR 峰面积比为纵坐标,建立零截距标准曲线,从而测定药品中GPC 的绝对含量。该方法简便快捷、准确可靠,物质鉴定和定量分析可以同步完成。
GPC作为一种手性药物,其两种对映异构体的生物利用度、代谢率,代谢产物以及毒性可能存在显著差异,所以测定GPC 产品的光学纯度是至关重要的。De Ferra 等[25]将GPC 的对映异构体与手性硼酸在二甲基亚砜中进行反应(图2),形成两种都含有两个手性中心的非对映体硼酸酯,通过1H NMR 光谱观察到这两种非对映体硼酸酯的胆碱甲基和苄甲基所对应的化学位移不同,从而根据相应特征峰面积大小计算出L-α-GPC 对映体的光学纯度。核磁共振谱法可用于GPC 的鉴定,也可用于测定最终得到的GPC产品的光学纯度。
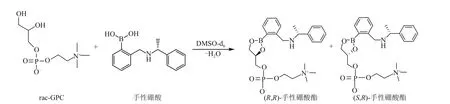
图2 手性硼酸与外消旋GPC的反应[25]
1.3 滴定法
《美国药典2019》[26]采用直接滴定法来检测从PC中提纯的GPC,0.1mol/L高氯酸的乙酸溶液作为滴定剂,采用电位法判断滴定终点,并计算消耗的滴定剂体积,用式(1)计算GPC的质量分数P。
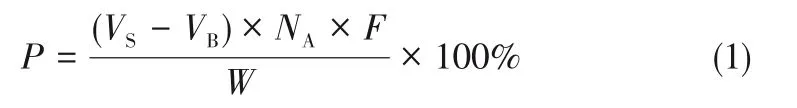
式中,VS为GPC 样品滴定达到终点时消耗滴定剂的体积,mL;VB为空白滴定终点消耗滴定剂的体积,mL;NA为滴定剂的标准浓度,mEq/mL;F为等效系数,257.2mg/mEq;W为样品质量,mg。并规定计算值在98%~102%为GPC的合格标准。
刘丹等[27]采用非水电位滴定法,将高氯酸标准溶液作为滴定剂,对GPC 样品进行滴定,计算滴加滴定剂的体积V和所测量的电压E,将一系列V、E值进行二级微商处理,用式(2)所示的内插法计算滴定剂的用量。

式中,Vφ为滴定终点时标准溶液的用量,mL;Va为二级微商为a时标准溶液的用量,mL;a为二级微商为零前的二级微商值;b为二级微商为零后的二级微商值;ΔV为二级微商为a至二级微商为b所加标准溶液的体积,mL。
采用二级微商法计算滴定剂的体积更为准确,求出的GPC 含量更为准确,但是此方法在制备GPC样品时用到了乙酸汞。黄祥元等[28]用乙酸代替乙酸汞溶液加入到GPC 样品中,这样既避免使用汞盐,所得到的滴定突跃现象也更明显。滴定法测量精度高,常用于提纯得到的GPC 产品含量的测定,但是其检测成本高、操作复杂,样品中少量的酸性物质也会引起误差。
1.4 液相色谱法
在定量分析GPC 含量时,高效液相色谱法是最常用的一种方法。刘狄等[29]采用高效液相色谱-蒸发光散射法(HPLC-ELSD),采用硅胶上键合1,2-二羟基丙基官能团的Diol 正相硅胶柱为分析柱,甲醇-水为流动相,检测了不同磷脂中PC 和GPC 的含量。但此方法的流动相中含有极性强的水,长期使用会对Diol正相柱造成损害,降低色谱柱的柱效,对检测结果有影响。杜章斌等[30]采用Alltech Silica色谱柱,甲醇为流动相,检测了PC水解液中的GPC 含量,此方法避免了极性强的水对色谱柱的伤害,增强了实验结果的准确性。
Kielbowicz等[31]开发了一种高效液相色谱-电喷雾检测器(HPLC-CAD)的检测方法,采用硅烷基配体官能化的球形硅胶作为固定相,乙腈-甲醇-10mmol/L 乙酸铵溶液作为流动相,此方法有效检测了PC、LPC 和GPC 的含量,但CAD 检测器的基线不平稳,杂峰很多,检测结果误差大。
为更准确检测PC 水解产物中GPC 及相关杂质的含量,《美国药典2019》[26]采用液相-示差折光法(LC-RID)和LC-ELSD 联用来检测提纯物中杂质的含量。LC-RID 是为了检测GPC 提取物中甘油的含量,色谱柱为L14 柱(10μm 硅胶化学键合强碱性季铵盐阴离子交换固定相),流动相为乙腈-水,测得甘油和GPC 的相对保留时间分别为0.6 和1.0,并且规定甘油含量小于0.5%为合格。LC-ELSD 法是为了检测GPC 提取物中的其余有机杂质,流动相按照以下方法配制,并按照表1中的时间进行梯度洗脱,可有效检测出GPC 提取物中各有机杂质的含量。
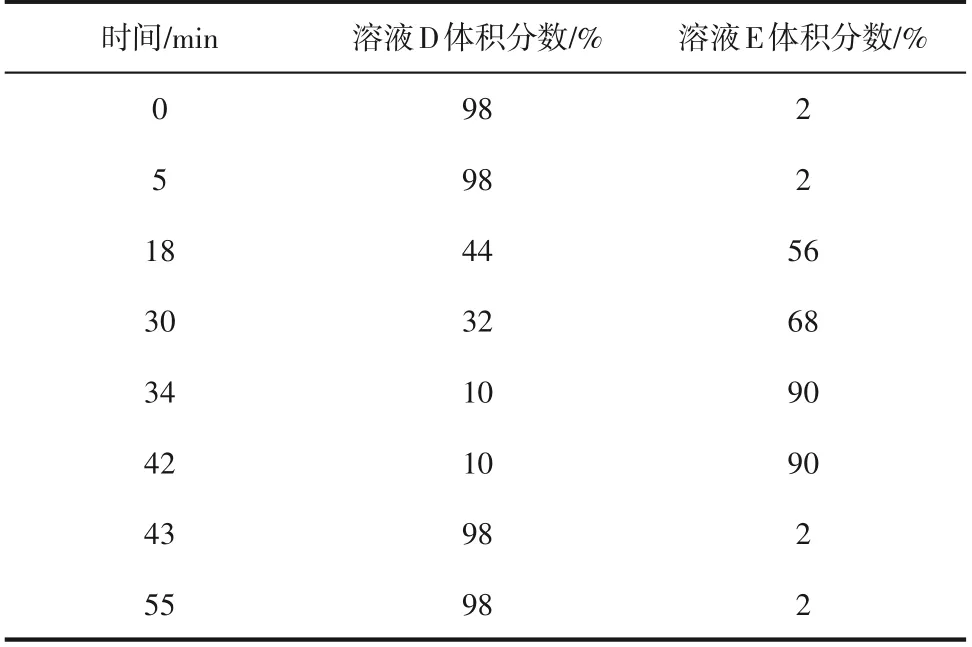
表1 流动相比例及其洗脱时间
为检测化学合成的GPC 及相关杂质的含量,傅超婷等[32]采用二元梯度洗脱程序,选取蒸发光散射检测器,以甲酸铵的乙腈溶液作为流动相对GPC待测样品进行第一液相色谱检测,用阳离子交换色谱柱,检测出杂质甘油磷酰肌醇(GPI)、氯化胆碱和甘油的含量;以乙腈-水为流动相对GPC待测样品进行第二液相色谱检测,用氨基键合硅胶色谱柱,检测出杂质甘油磷酰乙醇胺(GPE)的含量。这种方法能全面有效地检测GPC 样品中杂质的含量,能够更好地控制GPC 产品的质量。但此方法操作较为麻烦,需要切换两次色谱柱和两次流动相。陈东英等[33]利用极性硅胶柱作为色谱柱,甲醇-乙腈-缓冲盐溶液为流动相,采用示差折光检测器、蒸发光散射检测器或质谱检测器,能很好地检测出磷脂、胆碱、氯化磷酰胆碱(PC-Cl)和GPC的含量,并且各有关物质的分离度均大于1.5,峰形良好。液相色谱法是最常用的定量分析方法,可测定反应液中GPC 及其相关杂质的含量,还可以测定提纯后得到的GPC产品的纯度。
目前国内没有GPC 相关药物、保健品、食品和化妆品的生产,只有生产GPC 原料药的厂家,所以国内没有对GPC 的质量规定。国际市场上,用于药品、食品和保健品中的GPC 都是高纯度的GPC,其质量要求可参考美国药典。化妆品中添加的GPC暂时没有相关质量标准。
《美国药典》对GPC 的质量要求如下:①滴定法计算得到GPC 质量分数在98%~102%之间;②乙酸盐质量分数小于0.1%,氯化物质量分数小于0.02%,硫酸盐质量分数小于0.02%,磷酸盐质量分数小于0.005%、甘油质量分数小于0.5%。松醇、β-GPC、蔗糖、丝氨酸、GPE的质量分数都小于0.1%;③0.1g/mL 的GPC 溶液的旋光度在-2.4~-2.8 之间;④细菌总数不超过1000cfu/g,霉菌和酵母菌的总数不超过100cfu/g;⑤8.5g/100mL 或10g/100mL 的GPC 溶 液 的pH 在5.0~7.0 之 间;⑥GPC 固体水分含量小于1.0%,GPC 水溶液的水含量为14%~16%。
对各种分析方法的优缺点进行了比较分析,如表2所示。在反应过程中,可将薄层色谱法作为中控方法快速判断反应中是否有GPC 生成;进行定量分析时,液相色谱法是最常用的方法,可测定反应液中GPC 的浓度,从而计算反应收率。在分析提纯所得GPC 产品的质量时,其含量可用高精度的滴定法来测定,其相关杂质的含量可用液相色谱法测定。但没有一种检测器能检测出GPC 产品相关的全部杂质,对于缩水甘油、氯甘油这类有机杂质,需要RID 检测器来检测;对于GPE、GPI 这类杂质,需要ELSD检测器来检测。所以,多种检测器联用才能有效测定GPC 中相关杂质的含量,进一步判断GPC的质量是否符合要求。

表2 不同分析方法的比较
2 GPC的制备方法
GPC天然存在于生物物质中,所以早期是通过分离纯化技术从生物物质中提取GPC 的。1945 年Schmidt 等[2]便从牛的胰脏中萃取、纯化得到了GPC,并确定其结构由胆碱和α-甘油磷酸组成的。但是生物物质中GPC 含量极低,直接提取非常困难,产量很低。目前制备GPC 主要有水解磷脂类化合物和全化学合成两种方法。
2.1 水解磷脂类化合物
工业上采用水解法制备GPC 的原料为大豆磷脂粉末或蛋黄粉,两者的主要成分都是PC,PC的sn-1 脂肪酸酰基被水解得到sn-2-溶血磷酰胆碱(sn-2-LPC),再经过酰基自动转移得到sn-1-溶血磷酰胆碱(sn-1-LPC),最后水解掉剩余的脂肪酸酰基可得到GPC,其反应如图3。
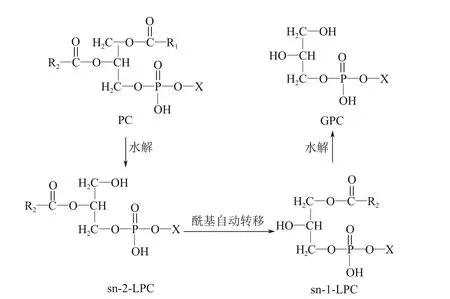
图3 PC水解的反应过程[34]
2.1.1 化学催化水解法
水解PC 常用的化学催化剂有金属钠、醇钠[35-36],化学催化水解的制备工艺简单,容易规模化生产,但GPC 的收率很低。PC 在离子交换树脂[37]的催化下醇解的效果较好,但离子交换树脂再生困难,并产生大量废水,不适合工业应用。为解决这些问题,Li等[38]考察了12种低沸点胺类催化剂的催化效果,发现在叔丁胺的催化下,PC 的转化率可达到98%以上,并且这类低沸点催化剂可以在旋蒸回收甲醇时被同步回收,可实现重复利用。
但是,这些化学催化剂毒性较大,最终得到的GPC 产品中仍然会有少量残留,使得GPC 产物的安全性存在问题。为了找到更好的催化剂,Li等[39]采用400℃下煅烧的硅酸钠作为催化剂,在甲醇中催化PC 的水解,PC 最高转化率能达到99.5%。由于硅酸钠无毒,易于从人体排泄,所以可以作为生产GPC的良性催化剂。
2.1.2 生物酶水解法
与化学催化剂相比,生物酶的毒性更小,也更安全,具备用量少、催化效率高和易于回收等特点。此方法得到的GPC 适合用于药物、食品和保健品等口服产品中,是PC水解反应中研究的热点。
Zhang 等[40]用磷脂酶A1(PLA1)作催化剂并加入辅助因子CaCl2增强PLA1的活性,GPC 的产率能达到94.5%。为进一步提高产率,Zhang 等[41]继续用Thermo4S-3 作为生物催化剂,并用响应面优化得到最佳反应条件,将GPC 的最大产率提高到96.8%。但是,在水相体系中,PC的溶解度非常有限,导致GPC 的生产效率很低,难以在工业生产中应用。为解决上述问题,Lu 等[34]使用Tween 20作为表面活性剂来改善PC 在水相中的溶解度和分散性,大大缩短了反应时间并提高了GPC的产量。
由于PC 在双相介质中的溶解度比在水中大得多,Bang 等[42]则考虑用双相介质来提高PC 的溶解度,以PLA1为生物催化剂在水-己烷的双相介质中催化PC 水解,使得GPC 的生产效率得到提高。但是,此方法的反应时间为30h,反应进行过于缓慢,不适用于工业生产。Kielbowicz等[31]研究发现,碱性pH 环境可以同时促进酰基迁移和水解过程,为了进一步提高PLA1的催化效率,Li等[43]成功开发一种简单的共沉淀方法将PLA1封装在金属表面活性剂纳米复合材料(MSNC)中,并利用碱性的2-甲基咪唑(2-Melm)对这种材料进行改性,得到催化活性增强的2-Melm@PLA1/MSNC 催化剂(图4)。这种复合材料具有碱性和两亲性,能够使酰基转移和酶水解在水-己烷的双相体系中高效有序地进行,并且具备良好的稳定性,重复利用不会影响其活性。
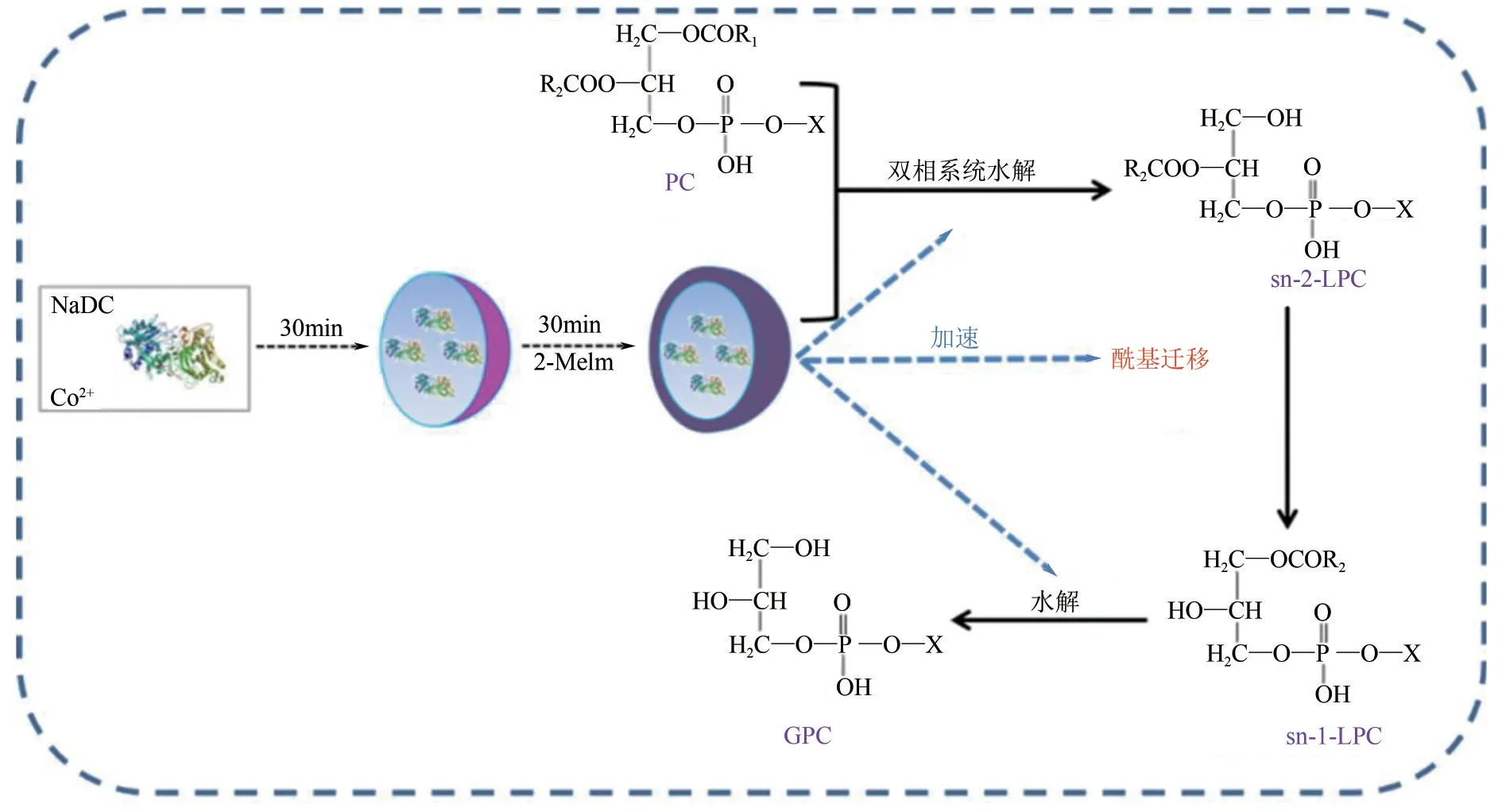
图4 2-Melm@PLA1/MSNCPC的合成及催化过程[42]
为了使GPC 的品质满足食品添加剂的要求,Kim 等[44]将大豆卵磷脂置于食品级萃取溶剂正己烷-水的双相介质中,选择Novozym435(诺维信脂肪酶435)催化其水解,过滤除去酶后所得滤液,通过双相介质的简单相分离即可得到纯度为98.6%的食品级GPC水溶液。
2.2 全化学合成法
缩水甘油或氯甘油与磷酰胆碱的盐反应生成GPC 的收率高(达到90%以上),反应工艺简单,是目前工业上最主流的合成路线。Park 等[19]以R-缩水甘油为原料,开发了两种合成GPC 的反应路线。路线1:R-缩水甘油先与苯甲醇进行开环反应,后经乙酰化、氢化,再与甲基苯磺酸胆碱和吡啶反应生成乙酰甘油磷酰胆碱,最后脱去乙酰基即得GPC。路线2:S-缩水甘油与甲基苯磺酸胆碱直接磷酸化得S-缩水甘油磷酰胆碱,再经过开环反应即得GPC。路线2 是在路线1 的基础上进行的改进,反应路线更短,操作更简单。
Song 等[45]利用环氧开环的原理,将R-缩水甘油与PC-Cl 在异丙胺的存在下进行亲核加成制备GPC,此方法制备工艺简单,但反应收率偏低,而且反应原料R-缩水甘油极不稳定,保存条件苛刻,增加了生产成本。Lee等[46]在此基础上,以R-氯甘油为原料,在低温条件下与NaOH的醇溶液反应生成R-缩水甘油,并立即将所制备的R-缩水甘油与PC-Cl以2∶1的物料比在50~60℃下反应制备GPC,将反应收率提高到70%~92%。Hwang等[47]利用此工艺路线,将450mmol PC-Cl溶解于甲醇中,依次加入900mmol 的KOH 和900mmol 的R-氯甘油,用一锅法制备出GPC,反应最大收率达到97%。此方法操作简单,中间产物少,适用于工业生产。PC-Cl的价格较为昂贵,通常不作为工业生产的原料。刘振等[48]则利用磷酰胆碱钙盐(PC-Ca)与碳酸钾水溶液反应制备磷酰胆碱钾盐(PC-K),将PC-K 与R-氯甘油反应制备GPC 粗品,此方法也避免用到不稳定的R-缩水甘油,原料的稳定性好且价格低,但是工艺操作比较复杂。尤家栋[49]利用R-氯甘油与NaOH 在乙醇中低温制备R-缩水甘油,过滤掉析出的盐后加入PC-Cl高温回流反应制备GPC,也具备很高的收率。
对各种制备方法的优缺点进行了比较分析,如表3所示。全化学合成法制备GPC具备收率高、产品纯度高、制备工艺完善的优点;但起始原料昂贵,最终得到的GPC 产品中也会残留一些基因毒性杂质,对产品的品质有严重影响。利用化学催化水解PC 同样存在这些问题,化学催化剂是毒性物质会影响产品在食品和药物中的应用。生物酶通常无毒无害,用生物酶水解PC 是制备食品级GPC 最好的方法,但此方法收率低,产品纯度低,对除杂的要求很高,规模化生产难度大。

表3 不同制备方法的比较
3 GPC的纯化方法
目前,市场要求GPC 产品的RID 纯度达到99.65%,旋光度在-2.4~-2.72,且单一杂质含量不能超过0.1%。但是,化学制备法会产生磷酰胆碱盐、缩水甘油、氯甘油和甘油等杂质,水解法会产生甘油磷酰乙醇胺(GPE)、甘油磷酰丝氨酸(GPS)、甘油磷酰肌醇(GPI) 等杂质。因此,GPC的纯化一直是一个难点。目前常用的纯化方法就是溶剂萃取法、结晶法和柱色谱法。
对于化学法制备的GPC粗品,刘振等[48]先用活性炭脱色,再用丙酮萃取除去其中的有机溶剂,最后用D001 大孔强酸性树脂和711 大孔强碱性树脂混床柱脱盐,可得到纯度为99.8%的GPC产物。丁建飞[50]采用GPC在无水乙醇中结晶的方法除去GPC中的有机杂质,得到的GPC纯度能达到99.5%。于振鹏等[51]考察了醇类、酯类、酮类和乙腈的结晶除杂效果,所得GPC 晶体的纯度都达到了99.5%以上。尤家栋[52]将反应液用丙酮萃取,然后用阴性离子交换树脂柱除去萃取液中的盐分,最后在无水乙醇中结晶得到精制GPC。
对于水解PC 得到的GPC 粗品,周丽等[53]采用D113 吸附树脂,将GPE 和盐类杂质除去,所得GPC纯度达到97%。Bang等[42]先将滤液用乙醚萃取三次以除去游离脂肪酸(FFA),接着用硅胶柱除去残留的FFA 和PC-Cl,从而得到纯化的GPC。Zhang 等[54]使用D001 阳离子和D301-111 阴离子交换树脂柱色谱法成功地将GPC 和GPE 分开,得到的GPC 纯度为98.8%。王志强等[55]将水解的PC 反应液直接进行氧化铝柱层析,再用D001 大孔离子交换树脂除去盐分,所得GPC纯度可达99%以上。
对各种纯化方法的优缺点进行了比较分析,如表4所示。在GPC的纯化中,目前最重要的还是结晶法和柱色谱法,结晶法收率低,操作较为复杂,可作为产品纯化的最终步骤;柱色谱法除杂效果好,但在除杂过程中会产生大量“三废”,纯化周期长,工业应用成本高。因此,开发适用于工业生产的除杂方法非常重要。

表4 不同纯化方法的比较
4 结语
人口老龄化是当今世界发展的一个重要趋势,老年人的精神健康问题也是全世界研究的热点,GPC能提高海马体中的乙酰胆碱水平,支持大脑和神经系统的功能,预防衰老造成的认知功能的下降,在中国这样的老龄化社会中具备良好的市场前景。
在目前的分析方法中,液相色谱法是GPC 的定量分析最常用的方法,液相色谱配合多种检测器联用还能检测各杂质的含量。薄层色谱法可作为中控方法,判断反应过程中是否有GPC 的生成。核磁共振谱法可用于GPC 的鉴定,也可用于测定最终得到的GPC 产品的光学纯度。滴定法测量精度高,常用于最终得到的GPC产品含量的测定。
在目前的制备方法中,全化学合成法制备GPC具备收率高、产品纯度高、制备工艺完善的优点;但起始原料昂贵,最终得到的GPC 产品中也会残留一些基因毒性杂质,对产品的品质有严重影响。利用化学催化水解PC 同样存在这些问题,这些化学催化剂同样是毒性物质,也会影响产品在食品上的应用。生物酶通常无毒无害,用生物酶水解PC是制备食品级GPC最好的方法,但此方法收率低,产品纯度低,对除杂的要求很高,规模化生产难度大。
在目前的纯化方法中,最常用的还是结晶法和柱色谱法:结晶法收率低,操作也较为复杂,可作为产品纯化的最终步骤;柱色谱法除杂效果好,但在除杂过程中会产生大量“三废”,纯化周期长,工业应用成本高。
在社会老龄化的背景下,GPC具有良好的市场前景。开发能规模化生产GPC 的工艺路线,适用于工业生产的除杂方法是其关键和难点。
