基于生物信息学的结直肠癌枢纽基因与预后相关基因筛选
2021-11-30李飞秦强强谷战峰张天祥申思周丽张乐莎
李飞 秦强强 谷战峰 张天祥 申思 周丽 张乐莎
结直肠癌是一种常见消化道恶性肿瘤,2020年全球癌症统计数据[1]显示结直肠癌新增病例250万,新增死亡90万,其发病率和死亡率居于肿瘤第三位和第二位[1]。近30年来,得益于以全结肠系膜切除术(CME)和靶向治疗等为代表的一大批新型治疗方法的出现,结直肠癌患者5年生存率已有较明显提高。但目前中国结直肠癌发病率和死亡率仍呈上升趋势并有年轻化倾向[2]。现有文献[3]表明包括遗传、环境和营养在内的多种因素参与结直肠癌的发生与发展,但其具体分子机制仍不清楚。结直肠癌现有的常用检测方法有结肠镜、电子计算机断层扫描(CT)、大肠潜血试验(FOBT)、粪便免疫学测试(FIT)等。其中最具代表性的结肠镜检查虽具较高特异度和灵敏度,但它为侵入性检查且价格高,检测前准备也较为苛刻,故患者接受性差。同时结直肠癌常以出血为临床表现,因症状无特异性,70%结直肠癌出血患者第一次就诊时被误诊为痔出血、息肉出血等。另外,现有以癌胚抗原(CEA)为代表的一类生物标志物特异度和敏感度并不理想[4],部分患者初次诊断即为中晚期,这是导致患者预后不良的一个重要因素。目前临床上对结直肠癌进行治疗和预后评价主要依据肿瘤TNM分期,该分期系统仅仅包含了肿瘤范围、区域淋巴结、远处转移三个宏观预测因素且未考虑患者个体特异性[5]。对于具体基因和信号通路研究还不充分,故利用生物信息学筛选高灵敏度和特异度生物标志物用于筛查、诊疗和预后预测就显得非常有必要。生物信息学是一门新兴学科,它结合生物学、数学和信息技术,使分析大型且复杂的分子数据集成为可能[6],本研究从基因表达综合数据库(GEO)筛选并下载数据集,通过比较肿瘤组织与相邻部位正常组织样本的基因表达谱筛选出差异表达基因(DEGs)。利用生物信息学方法分析出枢纽基因并使用GEPIA网站进行在线验证与预后分析,进而从分子角度探究结直肠癌发生与发展机制,为结直肠癌诊疗、预后预测及靶向药物研究提供理论依据。
材料与方法
一、基因芯片数据集来源
登录美国国家生物技术信息中心旗下GEO数据库(https://www.ncbi.nlm.nih.gov/geo/),并以colorectal cancer为关键词进行检索,筛选标准为:(1)数据集需来源于同一平台;(2)样本需包括结直肠癌组织与同一患者正常结直肠组织;(3)样本总量不小于20;(4)样本来源于“homo sapines”。共筛选出3套结直肠癌数据集(GSE110224、GSE41328、GSE22598),三套数据集均基于GPL570平台且为Affymetrix Human Genome U133 Plus 2.0 Array芯片。数据集GSE110224包含17个正常组织样本与17个结直肠腺癌样本,数据集GSE41328包含10个正常组织样本与10个结直肠腺癌样本,数据集GSE22598由17个正常组织样本与17个结直肠癌样本构成。
二、数据均一化与差异表达基因筛选
利用R语言4.0.3通过R软件“affy”包将原始表达数据批间差去除,随后进行背景校正,标准化处理,使用“limma”包筛选结直肠癌组织与正常组织差异表达基因,筛选条件:(1)校正后P值<0.05;(2)|log2FC|>1,其中log2FC>1为上调基因,log2FC<-1为下调基因。对三组数据集中均上调或者下调的基因取交集后使用R软件4.0.3“ggplot2”包绘制火山图。
三、差异基因富集与通路分析
DAVID数据库[7](https://david.ncifcrf.gov)集基因注释、可视化与综合发现为一体,是进行高通量基因功能分析的重要数据库。GO分析常用于功能富集研究,包含生物学过程(biological process,BP)、分子功能(molecular function,MF)和细胞组分(cellular component,CC)3类[7]。京都基因与基因组百科全书(Kyoto Encyclopedia of Genes and Genomes,KEGG)包含关于基因组生物途径和系统功能信息,可对基因功能进行系统分析。本研究使用DAVID数据库通过对差异基因进行注释并对其编码蛋白进行GO与KEGG分析,认为P<0.05具有统计学意义。
四、蛋白质相互作用网络构建与枢纽基因筛选
STRING数据库(http://string-db.org)涵盖了多种生物相关蛋白质—蛋白质关联数据,每个节点都代表一个基因、蛋白质或者分子,其间连线代表相互作用关系,可用于判断结直肠癌中差异表达基因编码蛋白质间相互作用。本研究将差异基因导入STRING数据库构建差异表达基因蛋白质—蛋白质相互作用网络(PPI),以综合分数(combined score)>0.4为筛选条件。所得结果导入Cytoscape v3.7.2软件进行可视化[7]。具有较高连通度(degree)的节点往往在对于该网络的稳定有着重要作用,因此使用Cytohubba插件筛选出PPI网络中连通度排名前10的基因并确定为枢纽基因。
五、枢纽基因验证与预后分析
GEPIA(http://gepia.cancer-pku.cn)数据库是由北京大学研制的在线分析网站,包含来源于TCGA的9 736个肿瘤组织与726个正常组织,以及GTEx数据库8 000余例正常组织数据。本研究包含其中300余例结直肠癌数据与712例正常组织数据[8-9]。通过GEPIA网站以枢纽基因相对表达量中位数为界值,根据差异基因mRNA表达量将患者分为高表达与低表达组进行组间比较,验证所筛选枢纽基因在正常组织与结直肠癌组织中表达差异与患者整体预后情况,认为检验值P<0.05为差异具有统计学意义。
结 果
一、结直肠癌差异基因表达
对GSE110224、GSE41328、GSE22598三组数据集进行标准化处理,处理结果显示三个数据集各样本表达量处于同一水平,表明其标准化特征良好。对三组数据集使用“limma”包筛选结直肠癌组织与正常大肠黏膜组织差异表达基因,共筛选出差异表达基因366个,其中明显上调基因128个,明显下调基因238个。样本均一化结果(图1)与火山图(图2)如图所示。

图1 数据集均一化处理。1A:GSE41328数据集均一化前;1B:GSE22598数据集均一化前;1C:GSE110224均一化前;1D:GSE41328数据集均一化后;1E:GSE22598数据集均一化后;1F:GSE100224数据集均一化后
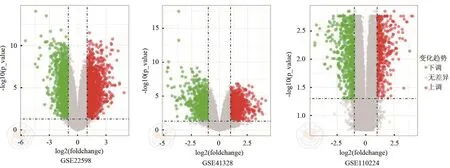
图2 差异表达基因火山图
二、差异表达基因的GO与KEGG分析
利用DAVID网站对366个差异表达基因进行GO富集分析与KEGG富集分析。GO分析结果表明差异表达基因主要参与蛋白水解、细胞黏附、细胞增殖的正性调节、炎症反应等生物学过程(图3A);主要定位于细胞外间隙、胞外区、蛋白质细胞外基质等细胞成分(图3B);主要参与锌离子结合、钙离子结合、受体结合等分子功能(图3C)。此外KEGG分析显示差异表达基因主要富集于细胞因子受体相互作用、趋化因子信号通路、PI3K-Akt通路等相关信号通路(图3D)。
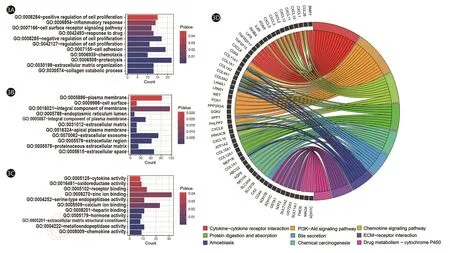
图3 差异表达基因的GO分析与KEGG分析信号。3A:通路1:生物学过程;3B:通路2:细胞组分;3C:通路3:分子功能;3D:KEGG信号通路富集图
三、PPI网络分析与枢纽基因鉴定
通过STRING数据库预测差异表达基因间相互作用,并将所得结果导入Cytoscape v3.7.2以构建蛋白质—蛋白质相互作用网络,得到一个包含282个节点和742条边的PPI网络。随后基于软件中Cytohubba插件筛选出连通度(具有较高联通度的节点意味着对维护整个网络的稳定更加重要)排名前10位枢纽基因,分别为CXCL8、CXCL1、SPP1、 CXCL12、 COL1A1、 SOX9、 MMP3、COL1A2、CD44和CXCL5。除CXCL12下调外,其余均为上调(表1)。
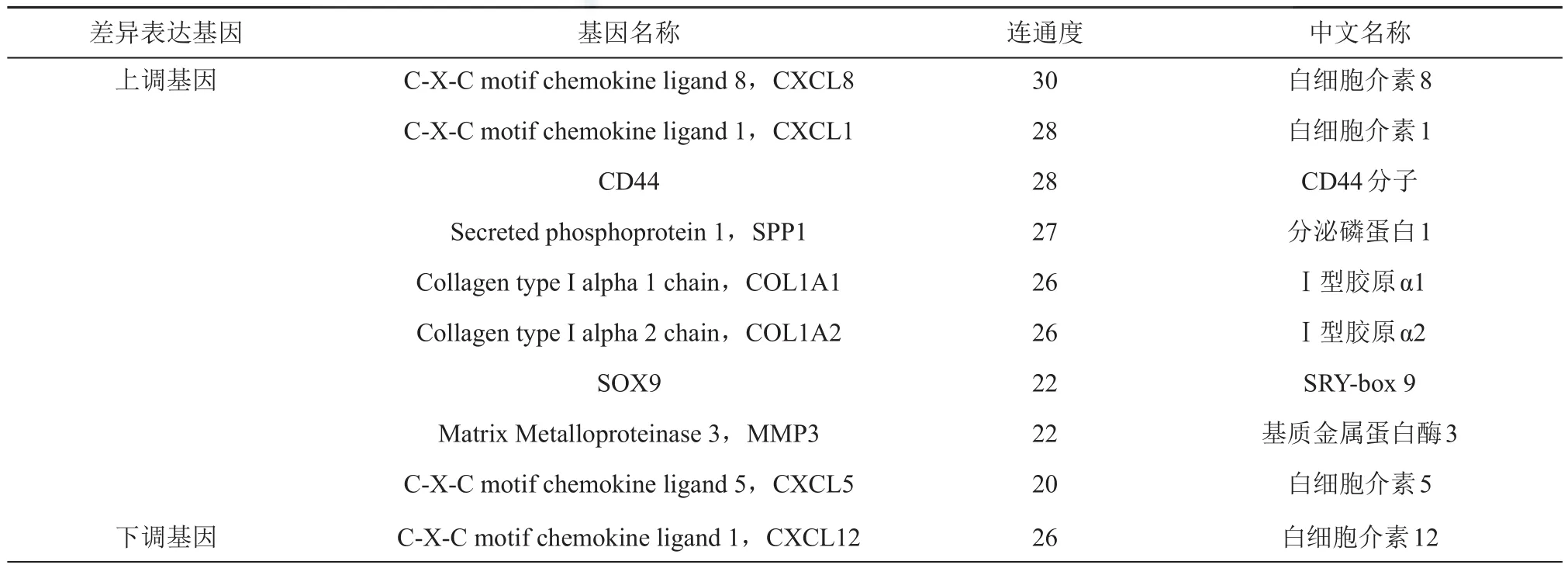
表1 10个枢纽基因
四、枢纽基因验证与生存分析
通过GEPIA(http://gepia.cancer-pku.cn)网站验证所筛选差异表达基因在正常人与结直肠癌患者中表达量,验证结果与GEO来源数据分析结果相一致。即在肿瘤组织中CXCL8、CXCL1、SPP1、COL1A1、 SOX9、 MMP3、 COL1A2、 CXCL5、CD44表达水平均较正常组织上调,CXCL12在肿瘤组织中较正常组织表达下调。除COL1A1和COL1A2以外的8个基因差异均具有统计学意义(LogrankP<0.05)(图4)。通过预后分析得出CXCL8、SPP1和COL1A2对于结直肠癌患者整体生存率、无病生存率均有影响(图5),提示其表达差异与患者预后相关。综合以上结果,显著性差异验证一致且与结直肠癌预后相关的基因为CXCL8和SPP1。

图4 枢纽基因在正常组织与肿瘤组织中的表达情况。T:Tumor,以红色表示;N:Normal,以灰色表示。COAD: Colon adenocarcinoma, 结 肠 癌 ; READ:Rectum adenocarcinoma,直肠癌

图5 预后相关基因与患者生存率的关系
讨 论
随着医学研究进入大数据时代以及多组学技术的发展,基于表达谱芯片和转录组测序的生物信息学分析在癌症发生发展机制探究、诊断和分型中应用越来越多[10]。其中不乏应用TCGA和GEO数据库进行分析的研究。GEO数据库为美国基因表达综合数据库,和癌症基因组图谱(The Cancer Genome Atlas,TCGA)数据库都是常用于进行生物信息学分析的数据库,且后者包含较为全面的癌症基因表达谱、突变基因谱,并配有相关临床信息,是目前最大的癌症基因信息数据库[11]。本研究对筛选出的GEO数据库中的三组结直肠癌表达谱基因芯片经过批间差去除、背景校正和标准化处理后,利用R软件对结直肠癌组织与正常组织进行分析筛选,得到366个差异表达基因,包括显著上调基因128个,显著下调基因238个。对差异基因进行GO富集分析发现,差异基因主要参与的生物学过程有蛋白水解、细胞增殖的正性调节、炎症反应等,差异基因主要定位于细胞外间隙、蛋白质细胞外基质等细胞成分。分子功能上与增强锌离子结合、钙离子结合作用相关。KEGG通路分析显示差异基因主要参与细胞因子受体相互作用通路、PI3K-Akt信号通路、趋化因子信号通路,表明炎症反应在结直肠癌中具有重要作用。
基于PPI网络筛选出的10个枢纽基因为CXCL8、CXCL1、 SPP1、 COL1A1、 SOX9、 MMP3、COL1A2、CXCL5、CD44,其中COL1A1、COL1A2两个基因在GEPIA验证差异表达时不显示具有统计学意义,可能跟这两种基因的基础水平表达量较高有关[10]。CXCL8是CXC家族典型趋化因子,具有负责招募和激活炎症细胞如中性粒细胞到炎症部位作用,CXCL8募集N2型TANs(肿瘤相关中性粒细胞),后者分泌的1型精氨酸酶通过抑制T细胞受体表达,减弱抗原特异性T细胞应答和募集T调节细胞从而引起免疫抑制,使机体免疫功能降低促进结直肠癌发生和发展;另一方面CXCL8也可通过下游胞内信号磷脂酰肌醇激酶诱导底物蛋白激酶B磷酸化,从而在调节肿瘤细胞存活、血管生成和迁移中发挥关键作用[12]。由此看来CXCL8在结直肠癌发生中可能为危险性因素,但我们的结果显示CXCL8在结肠癌患者中高表达,其下调为预后不良的表现,提示为保护性因素。对此我们认为有进一步验证的必要。另一趋化因子CXCL5可激活CXCR2 中 ERK/Elk-1/Snail途径和 AKT/GSK3β/βcatenin途径诱导上皮—间充质转化,并通过AKT/GSK3β/β-catenin/MMP7途径侵入CXCR2,导致肿瘤细胞迁移和侵袭[13]。CD44具有维持结直肠癌干细胞活性的功能,高表达CD44意味着预后不良[14]。COL1A1是Ⅰ型胶原a1,可通过WNT/平面细胞极性(PCP)途径激活三条通路Rac1-GTP、p-JNK、RhoA-GTP从而发挥不同作用,其中Rho GTPases和JNK途径将信号从细胞表面Frizzled和ROR2/RYK共受体传递到细胞核是肿瘤细胞转移的重要过程[15],因此,COL1A1可能参与了结直肠癌的转移过程。另一胶原COL1A2为Ⅰ型胶原a2,则通过调控细胞外基质(extracellular matrix,ECM)相关功能参与结直肠癌发生和转移,ECM是肿瘤微环境中最主要成分,其中Ⅰ型胶原在ECM中含量丰富,作为Ⅰ型胶原的一种,COL1A2可促进上皮—间充质转化从而增强肿瘤细胞对细胞凋亡抵抗,促进肿瘤细胞逃脱衰老过程[16];体外实验[13]结果也显示Ⅰ型胶原培养基上生长的结直肠癌细胞可通过诱导Cdx2瞬时转录下调导致分化受到抑制,这与COL1A2高表达患者预后不良相关。ECM与结直肠癌生物学行为密切相关,MMP3具有蛋白水解作用,能水解ECM,有利于结直肠癌细胞在降解的基质间隙及基底膜缺损处生长,同时其降解产物有化学趋化性并促进血管生成促进结直肠癌的侵袭和转移。失控的细胞增殖是结直肠癌细胞一种显著的生物学行为,体外研究[17]表明,上调的长链非编码RNA MALAT1通过下游靶基因miR-145使SOX9抑制作用减弱,导致SOX9表达上调,上调的SOX9使结直肠癌细胞分裂周期中停滞在G1期细胞比例减少,促进结直肠癌细胞增殖、迁移和侵袭。有报道[18]称,肠道中慢性炎症会增加结直肠癌的患病风险,在Rictor特异性缺失的结直肠癌患者中巨噬细胞对mTORC2抑制作用减弱,使骨桥蛋白SPP1分泌,导致炎症性结直肠癌的发生。
随后的研究中,我们使用GEPIA网站(包含TCGA数据库)对10个枢纽基因进行预后分析,发现CXCL8的低表达和SPP1、COL1A2的高表达与预后不良相关。关于结直肠癌,Chen等[17]曾应用TCGA和GEO数据库分析得出了207个结直肠癌差异表达基因,其中CXCL家族基因有四个亚型属于枢纽基因,而Gong等[18]也类似地发现CXCL家族的四个亚型与结直肠癌的预后相关。这些发现都与本文结论一致。本文也提出了COL1A两种亚型作为关键基因,其中COL1A2虽然GEPIA验证差异一致性中未显示出统计学意义,但与预后显著相关,在未来的研究中尚需进一步确认。
本研究为结直肠癌预后模型构建提供新的分子,从而进一步为结直肠癌诊疗与预后筛选提供理论依据。本研究也存在不足之处,结果虽经过不同网站和已有文献验证,但由于缺少具体实验数据,部分基因在结直肠癌发生发展以及预后等不同阶段作用未知,因此本研究中所涉及的分子及其机制仍需进一步分子生物学实验进行验证。
由于目前较多为单基因和针对二代测序进行的分析,广泛的探索性数据非常有限[19],今后的生物信息分析可能会向多基因模块发展。诸如加权基因共表达网络分析(WGCNA),通过对复杂的基因相互作用网络进行系统生物学方法分析,考虑同时评估所有基因的表达,以揭示共同表达(也可能是共同调节)的基因集群(模块)的变化,这样就可以认为是一种调控机制发生了改变而诱导出病因[20]。此外,基因水平分析得出的生物标记物也有一定局限性,由于基因转录和翻译水平的变化,使得仅使用基因组生物标记物是不够的,加之非遗传性因素的影响,可以考虑使用转录组分析来定义新的生物标记物对靶向药物的反应[21],并且多种RNA生物标志物的组合可以提高诊断和预后的敏感度和特异度[22]。
