Th17细胞的分化调控及其在炎症性肠病中的作用①
2021-11-26中国医科大学附属盛京医院消化内科沈阳110004
解 莹 李 岩 (中国医科大学附属盛京医院消化内科,沈阳110004)
炎症性肠病(inflammatory bowel disease,IBD)是一组慢性、复发性、炎症性疾病,主要包括克罗恩病(Crohn's disease,CD)和溃疡性结肠炎(ulcerative colitis,UC)。其中 UC 主要累及结肠,而 CD 可以累及从口腔至肛门的全消化道并且呈跳跃式分布。因IBD的复发性、致残性等特点,可严重影响人们的生活质量。既往IBD 的发病率以欧美居高,近年来IBD 在亚洲的发病率也逐年升高[1-3]。IBD 的发病机制尚未完全清楚,目前的研究认为可能与感染、环境、遗传、免疫、肠道微生物等因素有关,其中免疫功能异常在IBD的发病机制中日益受到关注。原始T 淋巴细胞在不同条件下可分化为辅助性T 细胞1(T helper 1 cells,Th1)、辅助性T 细胞 2(T helper 2 cells,Th2)、辅助性 T 细胞 17(T helper 17 cells,Th17)以及调节性T 细胞(regulatory T cells,Treg)等T 细胞亚群。既往研究认为,CD 主要表现为Th1 介导的免疫应答有关,UC 是Th2 介导的免疫应答有关[4-5]。近年来研究发现,Th17 细胞在 IBD 的发病过程中起到重要的作用。本文对Th17 细胞的分化调控及其在IBD中的作用进行综述。
1 Th17细胞分化及调控
Th17细胞是在2005年被首次提出的,以特异性分泌IL-17 为特点并且可以调节组织炎症的一类独立的辅助性T 细胞亚群。随着研究的深入,学者们逐渐揭示了Th17细胞的分化过程及其免疫功能,并且发现Th17 细胞在很多自身免疫性疾病中发挥着重要的作用[6]。
在STAT6 和T-bet 基因敲除的小鼠体内,给予IL-6 后可以单独诱导 Th17 细胞的分化[7]。原始 T 细胞分化过程中,高浓度的TGF-β 可促进其分化成Treg 细胞,低浓度的 TGF-β 与 IL-6 同时存在时可促进其向 Th17 细胞分化,并抑制 Th1、Th2 和 Treg 细胞分化,但在TGF-β基因敲除的小鼠体内Th17的数量随之减少[8-11]。说明 IL-6 和 TGF-β 的联合作用在原始T细胞向Th17分化过程中起到关键作用。
IL-23属于IL-12家族,由树突细胞、巨噬细胞产生。IL-23 并不影响原始T 细胞向Th17 细胞分化,但可以促进Th17 细胞的稳定与扩增,并且在IL-23缺乏时无法检测Th17细胞的存在[12-13]。IL-6可以上调 IL-23R、IL-6 和 IL-23,并依赖 STAT3 信号通路途径促进Th17 细胞的分化;同时STAT3 也可上调RORγt基因的表达进而促进Th17细胞的分化[14]。
IL-21 本身可由Th17 细胞分泌而产生,可通过其自反馈方式调节Th17 细胞的分化及功能。IL-21联合TGF-β可以独立诱导Th17细胞分化,但其效率低于 IL-6 联合 TGF-β[15]。IL-21 以依赖 STAT3 信号通路途径诱导Th17 细胞分化,还可以上调IL-17 和视黄酸受体相关孤核受体γt(retinoic acid receptor related orphan nuclear receptor gammat,RORγt)的表达,进一步促进 Th17 细胞的分化[16]。在 TNBS 及DSS 诱导的小鼠结肠炎中IL-21 高表达,IL-21 基因缺陷小鼠Th17细胞分泌的相关因子表达下降[17]。
RORγt 是Th17 细胞特异性转录因子。研究表明RORγt直接影响Th17细胞的分化,RORγt可诱导IL-17 和IL-17F 的基因转录[18];缺乏 RORγt 时,小鼠的自身免疫性疾病减轻,Th17 细胞明显减少;RORγt 和IL-6 同时缺乏的小鼠体内亦缺乏Th17 细胞。在RORγt缺陷的T 细胞不能分化成Th17 细胞,而增强RORγt 后可促进原始T 细胞向Th17 细胞分化[19]。IL-21 和 IL-23 可以通过 JAK-STAT 信号通路促进 RORγt 的表达,并且该途径需要依赖 IL-6[20]。RORγt 缺乏的 T 细胞其分泌 IL-17 和 IL-17F 的功能也下降[21]。
多项研究证实干扰素调节因子4(interferon reg⁃ulatory factor 4,IRF4)也可以干扰Th17细胞的分化。IRF4 基因敲除小鼠的 RORγt 的表达减少,Foxp3 的表达增多,原始 T 细胞不能分化为 Th17 细胞[22];IRF4 基因敲除后,IL-21 及 IL-17 的表达下降,同时RORα 和 RORγt 的表达下降,同时也证明 IL-21 对Th17 的自身正反馈调节有一部分是依赖于IRF4 途径实现的[23]。IRF4 可直接与 IL-17 启动子结合,上调RORγt和IL-17基因表达[24]。
正常情况下机体Th17 和Treg 细胞处于平衡状态,用以维持机体免疫应答和免疫稳态。Th17 细胞和Treg 细胞的分化都需要TGF-β,但分别依赖于不同的转录因子 RORγt 和 Foxp3[25]。低浓度的 TGF-β协同 IL-6 和 IL-21 促进 IL-23R 表达,促进 Th17 细胞分化。高浓度的TGF-β 抑制IL-23R 表达,促进Treg细胞分化。TGF-β 诱导的 Foxp3 抑制了 RORγt 的功能。IL-6、IL-21 和 IL-23 可减轻 Foxp3 介导的 RORγt的抑制作用。IL-2 和STAT5 在上调Foxp3 基因表达促进Treg 细胞分化的同时也抑制了Th17 细胞分化[26]。Foxp3 可通过直接抑制 RORγt 进而抑制IL-17A mRNA转录,从而抑制Th17分化及功能[27]。
TLR2 和TLR4 信号通路被激活后可促进Th17的增殖,抑制Treg细胞的分化[28]。TLR7信号通路的激活后可下调STAT3,进而抑制原始T 细胞向Th17细胞分化及IL-17的分泌[29]。
综上所述,Th17 细胞的分化受到多种细胞因子、转录因子以及信号转导通路的影响,具体机制见图1。
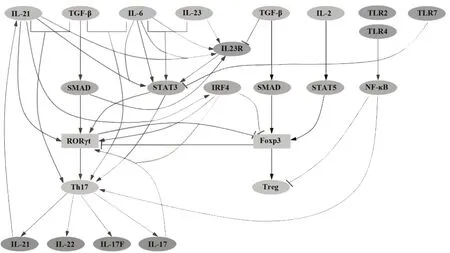
图1 Th17细胞的分化与调控Fig.1 Differentiation and regulation of Th17 cells
2 Th17细胞与IBD
Th17细胞主要分泌IL-17A、IL-17F、IL-21、IL-22及 IL-26 等细胞因子[30]。IBD 患者的外周血中 Th17细胞比例增加并且活动期高于缓解期[31]。
IL-17A 和 IL-17F 是 IL-17 家 族 成 员 ,主 要 由Th17 细胞分泌,然而 IL-17A 及 IL-17F 在 IBD 中的作用存在争议。活动期IBD患者血浆和肠黏膜均高表达IL-17[32]。活动期CD患者IL-17F的mRNA 表达水平明显升高[33]。IL-17A 和 IL-17F 与细胞表面受体IL-17RA、IL-17RC 结合,通过Act1、TRAF6 等进而激活NF-κB、MAPKs、C/EBP 等信号通路,促进炎症因子的释放,介导炎症反应。IL-17A 可与TNF-α 产生协同作用,促进TNF-α 所诱导的基因稳定性,增强IBD 的炎症反应[34]。IL-17A 可以募集和激活中性粒细胞,诱导趋化因子CCL20 的表达,促进其与CCR6受体的结合,以及炎症细胞的募集,增强肠黏膜免疫反应[35]。此外,参与IL-17A 信号转导的相关基因如 IL-23R、CCR6、Act1 等均与 IBD 有关[36]。由此可见,IL-17 作为Th17 细胞的主要效应因子参与IBD的发生发展,其机制可能为IL-17 促进炎症因子的释放,增强黏膜免疫反应,与其他炎症因子产生协同作用进而增强炎症反应等有关。针对抗IL-17A和IL-17F 的动物实验结果不尽相同。IL-17A 在疾病急性期和慢性活动期的作用不同,IL-17中和抗体可加重DSS 诱导的小鼠结肠炎,提示IL-17A 在小鼠急性结肠炎中具有保护作用[37];IL-17A 和 IL-17F 可诱发小鼠慢性肠道炎症[38]。小鼠接种IL-17 疫苗后,TNBS 诱导的结肠炎得到改善[39];但在另一项研究中,小鼠结肠炎加重且结肠中IL17 表达升高[40]。应用IL-17F 中和抗体后,小鼠DSS 诱导的结肠炎明显减轻[41]。目前临床上抑制IL-17 的药物主要有:IL-17 单抗:Secukinumab 和 Ixekizumab,IL-17RA 单抗Brodalumab,已被应用于银屑病、银屑病性关节炎、强直性脊柱炎的治疗。然而,针对抑制IL-17 治疗IBD 的临床试验研究结果也不尽相同。在Secukinumab 治疗活动性CD 的临床试验表明,中和IL-17A 并不能改善CD 病情,并且不良事件的发生率升高,并且,在接受Secukinumab 治疗风湿系统疾病的患者中,也有诱发新发IBD 疾病的报道[42-43]。IL-17RA 单抗Brodalumab 并不能有效控制中、重度活动期CD 患者的病情并产生不必要的副作用,可能与Brodalumab 不仅仅抑制了IL-17RA,同时还抑制了 IL-17 家族其他亚型有关[44]。Vidofludimus 和Tofacitinib 在IBD 的治疗中有一定的疗效,但是仍需要进一步更大规模的临床试验来评价其疗效及安全性,这两种IL-17小分子抑制剂不仅抑制了IL-17,同时也影响了 IFN-γ、IL-23、IL-6、JAK、STAT3 的表达[45-46]。综上所述,尽管 IL-17 在 IBD 患者中高表达,然而抗IL-17治疗目前看来并不是控制IBD的有效方法,IL-17 在IBD 中的作用是十分复杂的,有待更深入的研究。
IL-21 由Th17 细胞分泌,其可正反馈促进Th17细胞分化,促进IL-17 的分泌。在CD 患者的固有层CD3+淋巴细胞培养过程中阻断IL-21 的表达后,可下调 STAT3 和 IL-21[47]。IBD 患者高表达 IL-21,CD高于UC,在CD 病人固有层淋巴细胞中阻断IL-21后,下调磷酸化 STAT4 和 T-bet,抑制 IFN-γ,提示IL-21参与了CD的免疫反应过程[48]。
有研究指出,IL-22 对IBD 小鼠具有保护作用,通过NK细胞介导的天然免疫反应和Th17细胞介导的获得性免疫反应共同完成[49]。此外,IL-22可以促进结肠上皮细胞内STAT3 的活化,诱导了黏液相关分子STAT3 的依赖性表达和分泌黏液杯状细胞的修复[50]。尽管IL-22 可以在炎症反应高峰期发挥其抗炎的保护作用,但如果IL-22 与其受体结合后过度表达,也可促进结肠肿瘤的发生[51]。
研究显示,IBD 患者手术切除的结肠标本中IL-26 高表达,IL-26 通过 STAT1、STAT3、MAPKs 和PI3K/Akt 等信号通路途径诱导IL-6 和IL-8 的表达,参与IBD的病理生理过程[52]。
3 总结与展望
综上所述,Th17 细胞的分化受到诸多细胞因子、转录因子、信号通路等影响;并且在一定条件下不同T 淋巴细胞亚群细胞间存在相互促进、相互制约、相互转化的关系;尽管目前的研究显示Th17 细胞在IBD 的发生发展过程中主要起到促炎作用,但也有研究显示Th17 细胞分泌的细胞因子在一定条件下对IBD也起到保护作用。Th17细胞在IBD中的作用很可能是基于环境、不同细胞因子、不同信号通路等多种因素综合作用的结果。因此,深入探讨Th17 细胞及其分泌的细胞因子在IBD 中的作用机制,如何更好地调控Th17 细胞的分化及功能,使其向更有利于IBD 治疗的方向起作用,使其为IBD 的治疗提供新的治疗靶点,都有待进一步更深入的研究。
