WO3对Pt/α-Al2O3催化萘深度加氢的促进作用
2021-11-26梁瑜赵彤赵斌彬刘雷董晋湘唐明兴李学宽
梁瑜,赵彤,赵斌彬,刘雷,董晋湘,唐明兴,李学宽
(1 太原理工大学化学化工学院,山西太原030024; 2 中国科学院山西煤炭化学研究所,山西太原030001)
引 言
煤焦化过程产生大量的煤焦油,我国的煤焦油产能约2000 万吨/年[1]。目前,我国的煤焦油深加工技术仍处于相对落后水平,且在深加工的生产路线上还存在着不足,大部分都停留在了粗加工阶段[2]。萘是煤焦油中重要的芳烃化合物,含量为8%~12%[3],年产能约为200万吨。萘主要用来生产水泥减水剂和氧化生产苯酐,受石化原料新型减水剂和邻二甲苯氧化生产苯酐的冲击,萘下游产品严重滞销,造成萘的大量过剩,因此亟需将萘转化为高附加值的精细化学品[4]。十氢萘是优良的高沸点有机溶剂[5],可以作为超高分子量聚乙烯“干法纺丝工艺”的适宜溶剂,还可作为高超声速飞行器最佳的脱氢、裂解“吸热燃料”,利用萘加氢和十氢萘脱氢这一反应对可以使十氢萘作为新型储氢介质[6]。在高温条件下,十氢萘能够脱氢生成萘,而萘深度加氢过程是一个可逆的连串反应,其中萘加氢制四氢萘较为容易,受到高温脱氢反应的可逆性及芳环共振稳定性等因素的影响,四氢萘饱和加氢制十氢萘具有一定的难度[7]。因此,设计低温条件下高活性的加氢催化剂催化萘深度加氢对于十氢萘作为储氢介质工艺具有重要的意义。
目前芳烃加氢催化剂涉及过渡金属和贵金属催化剂,过渡金属多为Ni、Mo、Co 等金属硫化物负载于Al2O3制备得到,表现出较强的耐硫性,但也存在萘氢化程度低、操作条件苛刻等缺点[8-10]。与非贵金属基催化剂相比,铂族(Pt、Pd、Ru、Rh 等)贵金属催化剂因其深度加氢性能强、反应条件温和等优势已成为芳烃加氢领域研究的一大热点[11-12]。萘加氢反应的催化效率与活性组分的分散性和载体的性质密切相关。Liu 等[13]通过调控介孔ZSM-5 的Si/Al比来改变载体的酸性,发现负载型Pt/ZSM-5催化剂上萘加氢活性随载体酸量的增加而增加。Kumar等[14]制备了SiO2比例不同的Pt-Pd/SiO2-Al2O3催化剂,当SiO2比例为40%时,催化剂的酸性、金属分散性最佳,在180℃、20 MPa条件下实现了萘的完全转化,十氢萘选择性90%以上。Huang 等[15]采用共沉淀法制备了不同Al/B 比的硼酸铝贵金属负载催化剂,考察其对萘加氢的影响,当硼酸铝中Al/B 比为20 时,在220℃、5.17 MPa 条件下萘完全转化。Zheng 等[16]制备了不同SiO2/Al2O3比的Pd/USY 催化剂,随着SiO2/Al2O3质量比增加,Pd/USY 催化剂的B酸强度和酸量逐渐减少,萘加氢活性从90%降到20%。可以看出,通过调控沸石分子筛的骨架组成提高催化剂的酸性质有利于提高萘加氢活性,但由于分子筛孔道尺寸较小,分子扩散的限制致使反应条件比较苛刻,同时分子筛的成本也较高,限制了催化剂的发展。
WO3负载型催化剂,如WO3/ZrO2、WO3/TiO2具有优良的酸性质,在酸催化反应中具有重要应用[17-18]。Pt/WO3/MeOx催化剂具有好的催化加氢性能,一方面,氧化钨物种能提供足够酸量,另一方面Pt-WO3作为典型的金属-载体强相互作用体系强化了Pt 的分散和稳定性[19]。Kurosaka 等[20]采用顺序浸渍法制备了Pt/WO3/ZrO2催化剂,催化结果表明Pt-WO3提供了酸性位点和分散较好Pt 金属位点,在170℃、8 MPa 反应条件下对甘油氢解反应实现了24%的1,3-PDO 产率。García-Fernández 等[21-22]采用顺序浸渍法制备了Pt/WOx/Al2O3催化剂用于甘油氢解反应,研究发现WOx上的负电荷能使自Pt 表面溢流的H 上的电荷离域而形成Brønsted 酸位。WOx和Pt 的相互作用促使电子向W 转移从而降低了Pt 上的电子密度,提高了Pt 的稳定性和W 的还原能力,增强了溢流的氢生成氢质子的能力从而增加了体系的酸性[23-25]。WOx的负载量与存在状态是增强催化剂总酸性的关键,并且Pt-WOx间的电子相互作用也是提高反应活性的因素之一。
目前,关于载体Al2O3中引入Pt-WO3体系用于多环芳烃加氢反应的研究尚未见报道。基于Pt-WO3负载型催化剂的特点,本文以α-Al2O3为载体,其本身无酸性且结构非常稳定[26],利于深入理解WO3在反应过程中的作用。本文制备了系列不同WO3负载量的WO3/α-Al2O3,然后通过浸渍法负载Pt,合成负载型贵金属加氢催化剂Pt-WO3/α-Al2O3,系统研究了催化剂活性组分的粒径大小、酸性等特性,并将其用于萘低温加氢合成十氢萘反应。
1 实验材料和方法
1.1 所用化学试剂
六水合氯铂酸(H2Cl6Pt· 6H2O,≥98.0%),偏钨酸铵((NH4)6H2W12O40·xH2O,≥99%),萘(C10H8,≥98.0%),氧化铝(α-Al2O3,≥99.99%),正 癸烷(C10H22,≥98.0%),顺式十氢萘(C10H18,≥98.0%), 反式十氢萘(C10H18,≥98.0%),1,2,3,4,-四氢萘(C10H12,≥98.0%),均购自上海阿拉丁生化科技股份有限公司。
1.2 Pt-WO3/α-Al2O3催化剂的制备
1.2.1 WO3/α-Al2O3载体的制备 采用浸渍法制备了不同WO3含量的WO3/α-Al2O3载体:将载体α-Al2O3在110℃烘箱中干燥过夜;将定量的偏钨酸铵分别加入烧杯中并加入适量超纯水使其完全溶解;准确称取2 g 的α-Al2O3依次缓慢均匀地加入烧杯中,室温下匀速搅拌12 h;烘箱中110℃条件下干燥12 h,研磨得到固体粉末;将固体粉末样品均匀平铺在刚玉坩埚中,放入马弗炉,在空气气氛中以2℃/min 的升温速率升至500℃,恒温煅烧3 h。分别制备了6种xWO3/α-Al2O3(x为0,1%、5%、10%、20%和25%,质量分数,全文同)。
1.2.2 Pt-WO3/α-Al2O3催化剂的制备 采用浸渍法制备了Pt-WO3/α-Al2O3催化剂:将1 g 氯铂酸溶于50 ml 超纯水中备用;将xWO3/α-Al2O3载体在110℃烘箱中干燥过夜;取一定量的氯铂酸溶液和超纯水于烧杯中匀速搅拌一定时间后,将定量的载体缓慢均匀加入氯铂酸水溶液中,室温下匀速搅拌12 h;烘箱中110℃条件下干燥12 h,研磨得到固体粉末;将固体粉末样品均匀平铺在刚玉坩埚中,然后放入马弗炉,在空气气氛中以2℃/min 的升温速率升至300℃,在此温度下恒温3 h。根据氯铂酸的添加量,控制Pt的负载量为0.5%~2%。
1.3 分析测试仪器
采用日本理学Rigaku MiniflexⅡ型X 射线衍射仪(XRD),对样品的物相结构进行确认,CuKα(λ=0.15418 nm),扫描范围为5° ~ 80°;程序升温还原(H2-TPR)在麦克Auto ChemⅡ2920 化学吸附仪进行测定,用热导检测器(TCD)检测并记录谱图;样品在Tecnai G2 F20 S-Twin 型高分辨透射电子显微镜(美国FEI)上进行表征,在加速电压为200 kV 下测试样品铂纳米粒子的分散程度,利用粒径分布计算软件对催化剂样品中Pt 粒径进行统计。采用XPS(Axis Ultra DLD,Kratos,UK)对样品表面Pt 物种的价态进行了分析;吡啶吸附傅里叶变换红外光谱(Py-IR)在配备RT-DLaTGS 检测器的INVENIO R(布鲁克)光谱仪上记录;催化剂样品中负载的金属Pt、W含量通过电感耦合等离子体光谱法(Leeman Labs Profile ICP,Agilent 725 ICP-OES/Agilent 7700)分析测量;采用Jobin Yvon HR800 原位拉曼光谱仪在100~1100 cm-1范围内记录催化剂的室温拉曼光谱。
1.4 催化性能测试
萘加氢反应测试在25 ml 的高压反应釜(内含聚四氟乙烯内衬)中进行。将0.02 g 催化剂和5 ml的0.05 mol/L 萘的正癸烷溶液加入上述反应器中然后密封,用H2将反应器吹扫六次以排除空气,并将初始压力设定为3 MPa。将反应器以500 r/min 的搅拌速率快速加热至70℃,1 h 反应结束后,用冰水将反应釜迅速冷却至室温,分离催化剂,对液相产物进行气相色谱定量分析。采用日本岛津公司的气相色谱仪GC-2010 pro 对萘及其产物进行分析,氢火焰离子化检测器(FID),毛细管色谱柱,DB-5-HT(30 m×0.25 mm×0.25 μm),AOC-20i 自动进样器,分流比为50∶1,载气流速为1 ml/min;进样口温度为330℃,检测器温度为330℃,柱温在50℃保持2 min,以15℃/min的升温速率升到200℃保持一定时间,直至所有组分流出。
2 实验结果与讨论
2.1 Pt-WO3/α-Al2O3催化剂的表征
为了鉴定负载WO3和Pt 物种的分散状态和物相,对α-Al2O3负载WO3和Pt前后进行了XRD 表征,结果如图1 所示。可以看出,载体α-Al2O3在催化剂制备过程中物相保持不变,随着样品中WO3负载量的增加,晶化WO3的特征峰变强,当催化剂中WO3负载量为5%时,明显检测到了晶态WO3的形成(2θ=23.7°,33.8°,41.6°),且WO3特征衍射峰的强度随催化剂中WO3含量的增加而增加,表明α-Al2O3表面形成了较大颗粒的WO3物种。在样品负载Pt后[图1(b)],催化剂的XRD 谱图中未检测到铂的特征衍射峰(2θ=37.6°,45.8°,66.8°),WO3的特征峰没有发生变化。结果表明,在负载1%的Pt后,WO3/α-Al2O3结构保持稳定,而Pt以较高的分散状态沉积于载体表面。
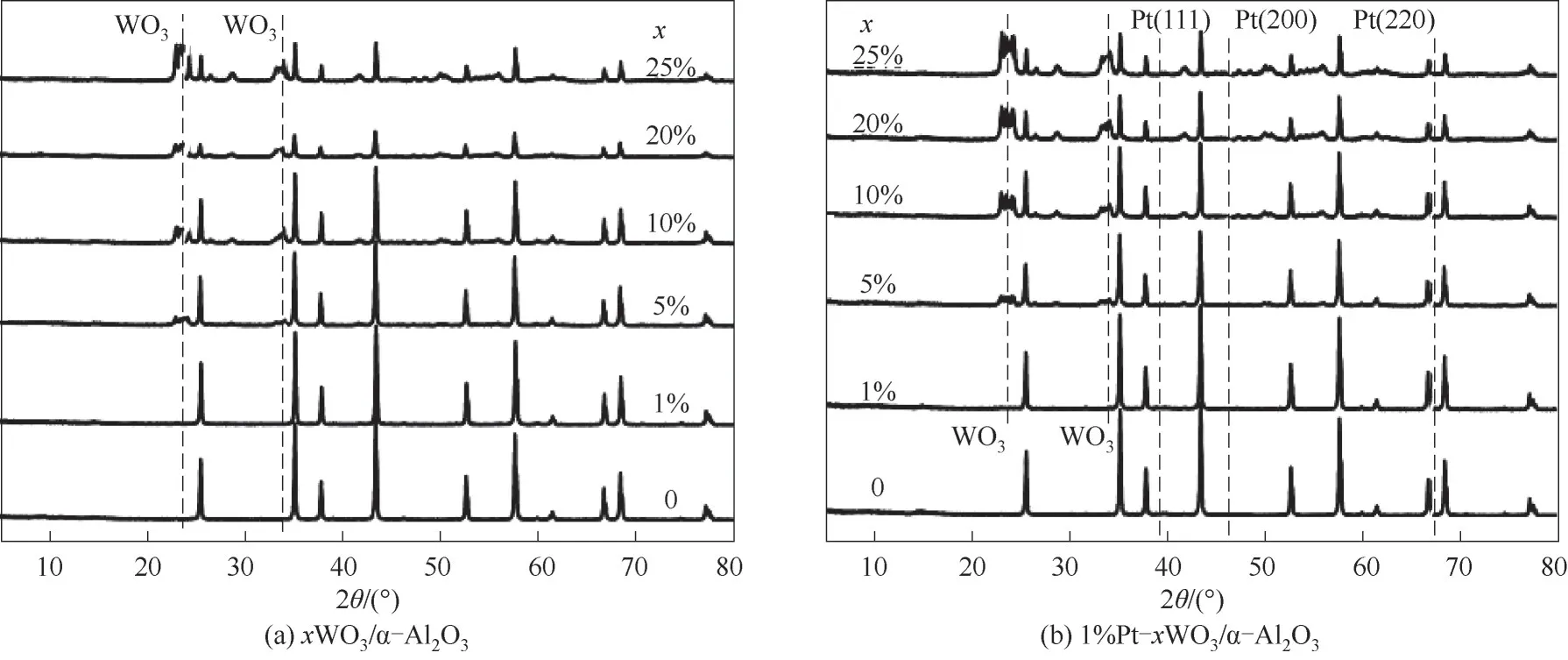
图1 粉末XRD衍射谱图Fig.1 Powder XRD patterns of samples
采用拉曼光谱表征了系列Pt-xWO3/α-Al2O3催化剂表面WO3物种的状态,并且以纯α-Al2O3和WO3作为对比,如图2 所示。由文献[27]得知,拉曼谱图中804、714、327、267、131 cm-1处的5 个拉曼谱带归属于晶态WO3的特征峰。结果表明,当WO3含量到5%时,明显检测到了晶化WO3的特征谱带,并且WO3特征峰的强度随催化剂中WO3含量的增加而增强,催化剂表面形成了具有较高结晶度的WO3物种,这与XRD得到的结果一致。

图2 样品的Raman谱图Fig.2 Raman spectra of samples
为了进一步研究确定Pt-WO3/α-Al2O3催化剂Pt、WO3物种的分散程度和粒径,进行了HRTEM 表征,同时研究Pt/α-Al2O3进行比较。如图3(b)所示,在1%Pt/α-Al2O3样品谱图中清楚地观察到Pt 纳米颗粒,平均粒径约10 nm,Pt-WO3/α-Al2O3催化剂[图3(a)]中能够观测到优先沉积在WO3附近约4.5 nm的Pt 颗粒,远小于1%Pt/α-Al2O3的颗粒尺寸。推测为由于WO3作为锚定位点限制了沉积Pt 物种的生长,导致沉积的Pt 颗粒尺寸变小,Pt 物种的分散性提升,1%Pt-20%WO3/α-Al2O3催化剂表现出较高的Pt 分散程度。同时进行了H2-O2滴定方法计算了样品Pt 分散度,结果表明Pt 分散度与WO3负载量相关,WO3的负载量为0、20%和25%时,Pt 对应的分散度分别为21.98%、55.9% 和34.9%,其中当20%WO3负载量时Pt的分散度最高。Pt的均匀分散和平均粒径的减小表明,在适宜的WO3负载量的情况下Pt 与WO3物种之间存在较强的相互作用,这是由于WO3中独特的缺电子环境所致[28]。

图3 催化剂HRTEM图Fig.3 HRTEM images of catalysts
通过程序升温还原(H2-TPR)进一步揭示Pt 与WO3物种之间的相互作用,表征结果如图4 所示。在1%Pt-20%WO3/α-Al2O3催化剂上观察到350、700℃两个H2还原峰。350℃附近出现的还原峰归属于正价态Pt物种的还原,700℃附近的峰归属于WO3的还原,与WO3/α-Al2O3相比,在负载Pt 后,1%Pt-20%WO3/α-Al2O3中WO3还原温度明显向低温移动50℃(约700℃处)。1%Pt-20%WO3/α-Al2O3相对低的WO3还原峰可能由于Pt 物种的氢溢流效应所致,催化剂中的Pt 物种优先沉积到WO3附近并与之发生相互作用,其中具有较高分散度的Pt有利于H2的快速解离,然后溢出到WO3表面促进了WO3物种的还原。上述结果表明,Pt 与WO3物种之间存在较强的相互作用[22,29-30]。
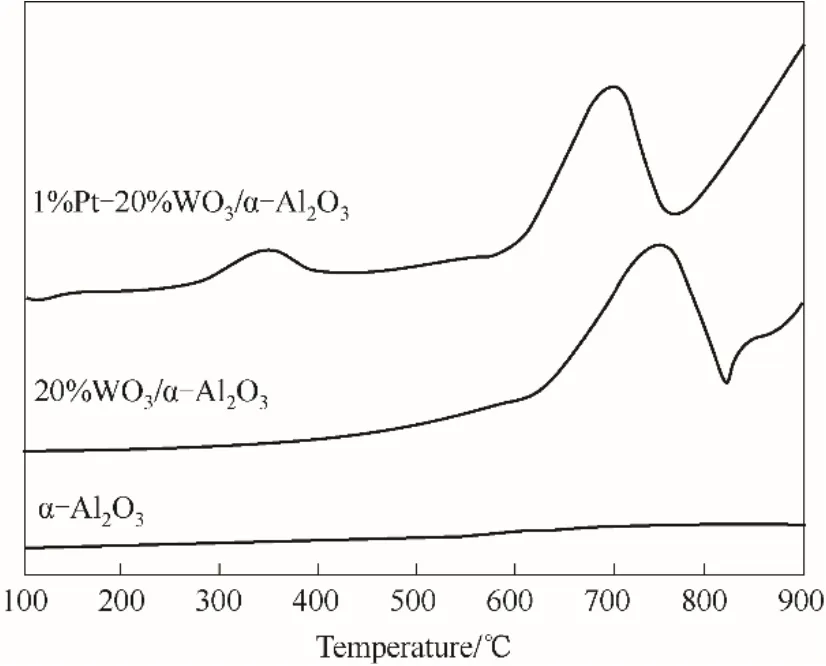
图4 α-Al2O3负载WO3、Pt前后样品的H2-TPR曲线Fig.4 H2-TPR profiles of α-Al2O3,20%WO3/α-Al2O3and 1%Pt-20%WO3/α-Al2O3 samples
采用XPS 光谱技术对1%Pt/α-Al2O3和1%Pt-20%WO3/α-Al2O3催化剂中Pt 表面化学价态进行了表征,结果如图5(a)、(b)所示。70~78 eV 的结合能(BE)区都呈现出宽广重叠的双重态,可以去卷积成72.9 eV 和76.3 eV 两个带,分别对应于Pt2+和Pt4+的4f7/2轨道。结果表明,Pt/α-Al2O3仅存在Pt2+物种,1%Pt-20%WO3/α-Al2O3检测到Pt2+和Pt4+物种,Pt4+物种的存在归因于Pt 的外层电子转移给缺电子状态的WO3物种。同时,与1%Pt/α-Al2O3催化剂相比,1%Pt-20%WO3/α-Al2O3催化剂中Pt2+结合能位置向更高的结合能方向移动。同时分别对10%、20%及25%WO3负载量的1%Pt-WO3/α-Al2O3催化剂中表面钨物种的化学价态进行了表征,如图5(c)~(e)所示。其中35.1 eV 和36.1 eV 两个谱带分别对应于W5+和W6+的4f7/2轨道,通过对应的峰面积进行计算,WO3负载量从10%增加到20%,W5+物种含量由9.8%增加至22.7%。表明在中等WO3负载量下,W5+含量与负载量成正比关系,缺电子状态下的W5+物种对Pt 有很好的锚定作用,有利于Pt 物种的分散。25%WO3负载量下WO3在α-Al2O3表面聚集生长为较大的粒子,W5+物种含量仅占5%,较少的锚定位点导致Pt 物种更容易团聚,这与H2-O2滴定获得的样品Pt 分散度结果一致。上述结果说明在1%Pt-20%WO3/α-Al2O3中,Pt 与WO3之间发生相互作用,Pt 物种电子向WO3转移,WO3的存在对Pt 物种的稳定和分散起到了促进作用[31-32]。分散度较好的Pt 有利于提高H2的解离和溢出,最终1%Pt-20%WO3/α-Al2O3表现出了更高的萘加氢催化性能。通过XPS对典型的催化剂(1%Pt-20%WO3/α-Al2O3)表面元素进行了分析,如图5(f)所示。XPS 全谱分析中可以检测到Al、W、Pt 和O 元素,未检测到Cl 元素的特征峰(约200.5 eV),表明Pt 前体中的氯离子经煅烧处理后大部分Cl 被脱除,残留的Cl 含量非常低,低于XPS仪器的检测下限(<0.1%)。

图5 催化剂的Pt 4f、W 4f XPS和XPS全谱分析Fig.5 XPS spectra of Pt 4f and W 4f of catalysts
催化剂表面酸性是影响萘加氢催化剂加氢性能的关键因素,采用Py-IR 以考察催化剂中的表面酸位,结果如图6 所示。Py-IR 谱图中,1452 cm-1和1612 cm-1的吸收带归属于Lewis 酸位,1545 cm-1的吸收带归属于Brønsted 酸,1486 cm-1处的谱带表明吡啶分子既与Lewis 中心又与Brønsted 酸性中心配位[33-34]。对所有负载WO3后的样品,其Lewis 酸位的特征吸附带非常明显,故催化剂主要由Lewis 酸中心组成,随着催化剂中WO3含量的增加,Lewis 酸位的数量呈现增加趋势,结合XRD 及Raman 表征结果可以证实,催化剂表面Lewis 酸位主要是由WO3纳米团簇提供,且酸量与WO3的含量成正比。催化剂的表面酸性有利于萘分子的吸附,表面中等密度分布的WO3(<20%),有助于富含电子的萘环在催化剂表面快速吸附反应,从而促进了对萘的深度加氢性能,继续提高WO3负载量(25%),由于催化剂表面酸量过高对萘分子产生了强的吸附能力,不利于氢气的解离和产物的脱附,从而造成催化活性的降低。

图6 1%Pt-xWO3/α-Al2O3的Py-IR光谱Fig.6 Py-IR spectra of 1%Pt-xWO3/α-Al2O3 catalysts
2.2 催化剂萘加氢性能评价
2.2.1 催化活性和选择性 在低温反应条件下(70℃),考察了合成的6 种不同WO3负载量催化剂的萘加氢性能,反应结果如图7 所示。对于1%Ptα-Al2O3催化剂,萘的转化率仅为91.6%,负载WO3后的催化剂转化率均达到了100%。且随着催化剂中WO3含量由1%增加到20%十氢萘的选择性从10.2%增加到100%,其中顺式十氢萘与反式十氢萘占比分别为80%与20%。而随着催化剂中WO3含量继续增加到25%时,十氢萘的选择性下降到64.0%。1%Pt-20%WO3/α-Al2O3催化剂具有最佳的催化性能,萘的转化率和十氢萘的选择性达到100%。上述结果表明,Pt纳米颗粒作为萘加氢的催化活性中心,1%Pt-20%WO3/α-Al2O3催化剂上Pt 纳米颗粒具有最高的分散度且粒径较小,WO3和Pt 纳米粒子间强相互作用有助于Pt 物种的分散,产生了更多的Pt-O-W 界面,促进了氢溢流的发生,有利于萘快速加氢反应[27]。同时Pt-Al2O3催化剂中引入WO3可以提供更多的酸性位点,更容易对富含电子的萘环产生强的吸附作用[13],有助于提高萘的催化反应活性。对于载体表面中等密度分布的WO3,负载量的增加造成酸量的增加,从而实现对萘加氢性能的促进,25%WO3负载量下,催化剂具有较高的酸量,对萘分子的吸附能力过强,难以脱附,使得萘深度加氢性能下降,未能完全转化为十氢萘。
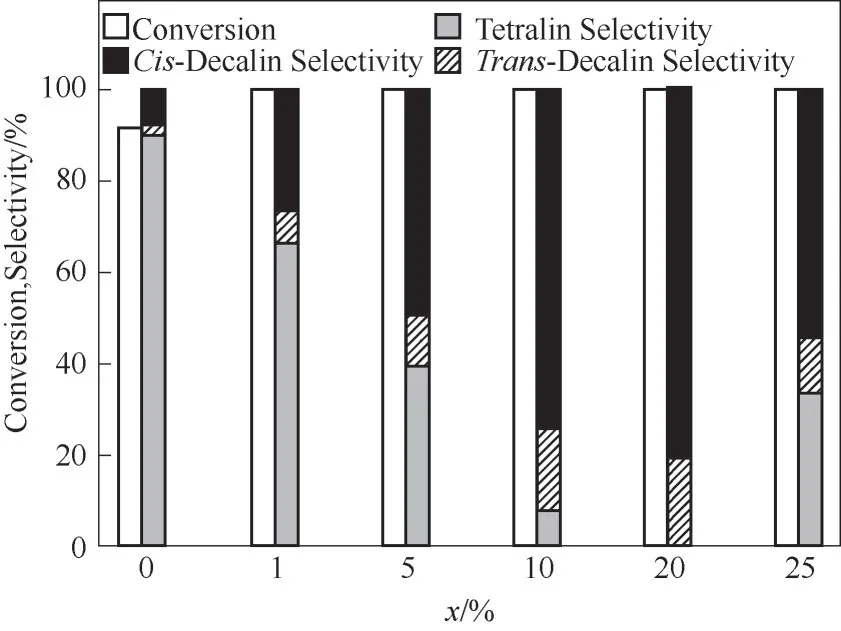
图7 萘在1%Pt-xWO3/α-Al2O3催化剂上加氢反应结果(反应条件:0.05 mol/L萘溶液5 ml,催化剂0.02 g,初始H2压力3 MPa,反应温度70℃,时间1 h)Fig.7 The reaction results for naphthalene hydrogenation over 1%Pt-xWO3/α-Al2O3 catalyst(reaction conditions:5 ml naphthalene solution of 0.05 mol/L,0.02 g catalyst,3 MPa initial H2 pressure,temperature of 70℃,and time of 1 h)
2.2.2 Pt 负载量的影响 催化剂中Pt 作为氢解离活性中心,通常其负载量与催化活性成正比,在3 MPa H2和70℃、1 h 反应条件下,考察了Pt 负载量(y)对萘加氢催化性能的影响,反应结果如图8 所示。由图可知,当Pt 的负载量为0.5%时,萘的转化率为59.36%,加氢产物以四氢萘为主,这主要归因于萘在催化剂活性中心的吸附能力远高于四氢萘,当催化剂上Pt 含量较低时,加氢活性位点较少,导致萘加氢速率降低,故在相同反应时间内产物以四氢萘为主,这与后续温度对反应结果的影响所得的结果一致。当Pt 负载量增加至1%以上时,萘的转化率和十氢萘的选择性均为100%,反应产物中没有检测到其他萘开环、十氢萘开环及断链产物,表明低温反应条件下可以抑制多环产物在酸性催化剂上的开环反应。研究结果表明,Pt-WO3/α-Al2O3催化剂具有高的产物选择性,提高Pt 的负载量可以显著地提升萘加氢催化效率,有助于萘深度加氢合成十氢萘。
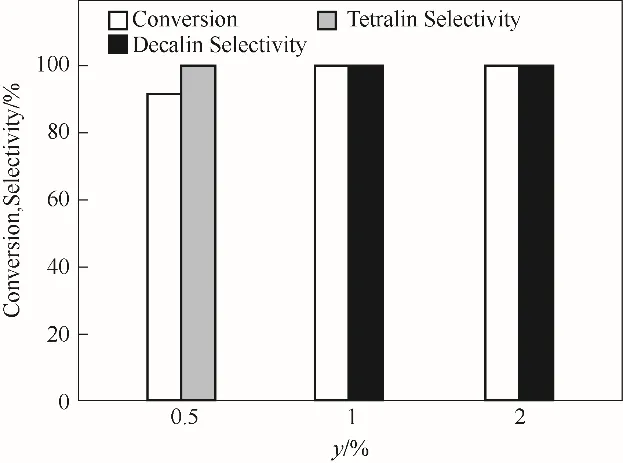
图8 yPt-20%WO3/α-Al2O3催化剂上萘加氢反应结果(反应条件:0.05 mol/L萘溶液5 ml,催化剂0.02 g,初始H2压力3 MPa,温度70℃,时间1 h)Fig.8 The reaction results for naphthalene hydrogenation over yPt-20%WO3/α-Al2O3 catalyst(reaction conditions:5 ml naphthalene solution of 0.05 mol/L,0.02 g catalyst,3 MPa initial H2 pressure,temperature of 70℃,and time of 1 h)
2.2.3 反应温度的影响 以1%Pt-20%WO3/α-Al2O3为催化剂,考察了不同反应温度对萘加氢催化性能的影响,反应结果如图9所示。可以看出,随着反应温度从30℃升高到70℃,萘的转化率从10.18%增加到100%。当反应温度在30~50℃范围,反应产物主要为四氢萘,十氢萘比例极少(约1%);反应温度在60℃以上时,萘全部转化,十氢萘的选择性开始上升,直到反应温度达到70℃以上时十氢萘的选择性达到100%。在萘和四氢萘共存的体系中,与四氢萘相比,萘具有较高的电子密度,致使萘在催化剂上具有更强的竞争吸附作用,从而萘优先在催化活性中心上完成加氢反应,然后四氢萘再发生吸附和加氢[35]。结果表明,反应温度的升高可以显著地提升萘加氢催化效率,有助于萘深度加氢合成十氢萘。
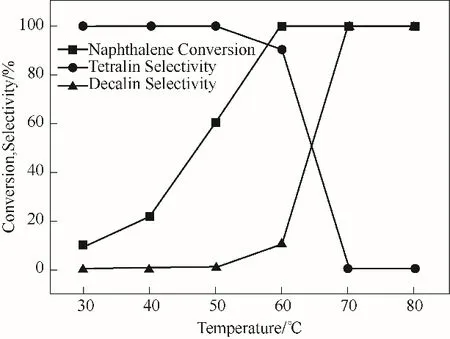
图9 1%Pt-20%WO3/α-Al2O3催化剂在不同反应温度下萘加氢的反应结果(反应条件:0.05 mol/L萘溶液5 ml,催化剂0.02 g,初始H2压力3 MPa,时间1 h)Fig.9 Catalytic results for naphthalene hydrogenation over 1%Pt-20%WO3/α-Al2O3 at various reaction temperature(reaction conditions:5 ml naphthalene solution of 0.05 mol/L,0.02 g catalyst,3 MPa initial H2 pressure,and time of 1 h)
2.2.4 稳定性实验 在优化反应条件下,对1%Pt/20%WO3/α-Al2O3催化剂进行了4 次循环利用实验,结果如图10 所示。循环实验表明催化剂具有良好的稳定性,经过4 次循环后萘的转化率仍为100%,表明加氢反应中铂作为催化反应活性中心,具有较好的稳定性和重复利用性,同时也印证了催化剂中残留的Cl 对催化剂活性及稳定性的影响较小。深度加氢产物十氢萘的选择性略有下降,可能归因于催化剂回收过程中的损失和催化剂反应过程中结构的微小变化。

图10 1%Pt-20%WO3/α-Al2O3催化剂4次循环使用反应结果(反应条件:0.05 mol/L萘溶液5 ml,催化剂0.02 g,初始H2压力3 MPa,时间1 h)Fig.10 Reusability of the 1%Pt-20%WO3/α-Al2O3 catalyst for naphthalene hydrogenation(reaction conditions:5 ml naphthalene solution of 0.05 mol/L,0.02 g catalyst,3 MPa initial H2 pressure,and time of 1 h)
由于制备的催化剂在反应前未经氢气预处理,对第一次反应后1%Pt-20%WO3/α-Al2O3催化剂中W 与Pt 物种的化学价态进行了XPS 表征分析,结果如图11 所示。经第一次加氢反应后催化剂中71.5 eV 的谱带归属于Pt0的4f7/2轨道,表明在加氢反应的过程中正价态的Pt物种被原位还原成Pt0,Pt0能有效地解离H2物种,从而实现萘的深度加氢。对W 物种的XPS 表征结果表明,35.3 eV 和36.1 eV 两个谱带分别对应于W5+和W6+的4f7/2轨道,对应的峰面积计算得到W5+物种含量为20.9%,与反应前催化剂相比W 物种价态几乎未发生变化。表明加氢反应中原位生成的零价Pt 作为催化反应活性中心,具有较好的稳定性和重复利用性,初始催化剂中残留的Cl 可能在反应气氛下在Pt 的还原过程中被脱除,其对催化加氢反应活性影响较小。

图11 第一次反应后1%Pt-20%WO3/α-Al2O3催化剂XPS谱图Fig.11 XPS spectra of Pt 4f and W 4f for 1%Pt-20%WO3/α-Al2O3-Used catalysts
3 结 论
通过浸渍的方法,以偏钨酸铵为钨源制备了系列不同WO3负载量的Pt-WO3/α-Al2O3催化剂。实验结果表明,与1%Pt-α-Al2O3相比,1%Pt-20%WO3/α-Al2O3催化剂表现出较高的萘加氢催化活性和十氢萘的选择性(100%)。在无酸性存在的α-Al2O3载体中引入WO3可以提供酸性位点,酸性位点对富含电子的萘环产生强的吸附作用,同时WO3与Pt 产生强相互作用,有助于Pt 物种的分散并促进了氢溢流的发生,有利于萘快速加氢反应,有利于Pt-WO3/α-Al2O3催化剂在温和条件催化萘深度加氢到十氢萘,展现出良好的催化活性。
