基于知识学习挖掘乳腺癌与甲状腺癌的共享功能模块和核心基因
2021-11-18许德华陈晓琳廖苑君蓝树金孙胜南饶绍奇
许德华,陈晓琳,廖苑君,蓝树金,孙胜南,李 让,饶绍奇
(广东医科大学公共卫生学院1,医学系统生物学研究所2,广东 东莞 523808)
乳腺癌(breast cancer,BC)和甲状腺癌(thyroid cancer,TC)是我国常见的女性恶性肿瘤。BC 年发病数约为30.4 万,位于女性恶性肿瘤发生首位[1]。TC年发病数约为15.10 万,位于女性恶性肿瘤发生第4位[1]。BC与TC 的发病率都呈逐年上升趋势[2,3],且根据以往研究报道[4],TC 或BC 患者发生第2 原发BC或TC 的风险相对更高。我国2001-2010 年调查研究发现[5,6],患BC 的女性未来患TC 的可能性是普通人的2 倍,而患TC 的女性未来患BC 的几率比普通人群高67%。研究显示[7,8],BC 患者一级亲属患TC的风险更高,提示BC 和TC 之间可能存在某种家族联系。有研究显示[9],BC 和TC 这两种癌症都受下丘脑-垂体-腺体轴调控,与性别、环境、药物治疗以及遗传等因素有关,但根本原因尚未明确。因此,本研究基于基因多效性理论,利用转录组数据结合生物学网络分析方法挖掘两种疾病的共享功能模块及核心致病基因,为揭示两种癌症发病的关联机制提供新思路,现报道如下。
1 资料与方法
1.1 数据来源 从TCGA 数据库(https://tcga-data.nci.nih.gov/tcga/)中分别下载BC 和TC 的RNA-seq数据和临床资料。样本剔除标准:①重复测量样本;②临床信息缺失样本。异常表达基因剔除标准:表达值在80%的样本中均少于0 的基因。生物分子网络的构建利用人类蛋白质参考数据库[10](Human Protein Reference Database,HPRD)中的蛋白质互作关系(protein-protein interaction,PPI)。
1.2 共享风险基因的筛选 利用“edgeR”R 软件包对BC 和TC 的RNA-seq 数据分别进行差异分析,以FDR 校正后P<0.05 且|logFC|>1 为标准筛选差异表达基因(differentially expressed genes,DEGs)。最后,利用韦恩图对BC 和TC 的DEGs 按共同上、下调取交集,得到两种癌症的共享风险基因(common risk genes,CRGs)。
1.3 风险基因网络的构建 以CRGs 为初始种子基因,当任意两个基因在HPRD 存在互作关系,那么视为它们在网络中存在连接,网络的节点代表着基因(包含种子基因及其一级邻居基因),最终构建出一个无向的共享风险基因网络。在Cytoscape 软件(版本3.6.1)进行网络可视化。
1.4 BC与TC 关键功能模块的挖掘 利用Newman网络分割算法[11,12]进行功能模块的挖掘,此算法的核心是将网络分解问题转变成二次型优化问题,即求使目标函数Q 最大的列向量s 所对应的数值:

其中B 是一个对称矩阵,表示为

s 为一个列向量,可表示为

si或sj是指节点i 或j 所属的子网;sT表示s 的倒置,表示行向量;A 为s 内所有节点的邻接矩阵,反映基因是否存在互作关系,当Aij=1 表示,i 基因和j 基因互作,Aij=0 则无。m 为网络中边的总数,ki或kj为节点i 或j 的连通度。根据柯西(Cauchy)不等式,s 取B 最大特征值对应的特征向量,使目标函数的使Q 达到最大。根据s 取值符号方向将网络切割成两部分,重复这一步骤,至网络无法分解为止。最后将节点数大于50 的子网作为模块。以上计算过程在R 语言(version3.6.1)中进行。
1.5 拓扑学分析及模块核心基因识别 利用网络直径、特征路径长度、连通度、无标度网络特性等指标对生物分子网络进行拓扑学分析。基于泊松分布,通过识别某个基因的连通度是否显著大于一般基因进行连通度(k)的检验来筛选核心基因:

其中,λ=NP,λ 表示节点的期望连通度;N 为节点数目;P为节点间发生连接的概率。以Bonferroni校正后P<0.05 为差异有统计学意义。
1.6 功能富集分析 利用“ClusterProfiler”R 软件包[13]对关键功能模块进行KEGG 通路富集分析。设定FDR 校正后P<0.01 为统计学意义显著。对各模块富集到的通路经KEGG 数据库进行分类,探讨关键功能模块之间的关联。
1.7 生存分析 利用“survival”R 软件包中的Kaplan-Meier 法绘制生存曲线对核心基因进行生存分析,以P<0.05 为差异有统计学意义。
2 结果
2.1 共享风险基因的筛选 对TCGA 数据集进行预处理后,得到BC 样本1036 个(病例924 个+正常112 个);TC 样本500 个(病例442 个+正常58 个)。差异分析后,在BC 中有5289 个DEGs(3200 个上调,2089 个下调),TC 中2972 个DEGs(1926 上调,1046 下调)。将BC 和TC 的DEGs 按上下调取交集后得到1136 个CRGs(708 个上调,428 个下调),见图1。

图1 共享风险基因韦恩图
2.2 BC 和TC 共享风险基因网络的构建 基于HRPD 数据库的PPI,构建出一个包含2856 个节点和4408 个互作对的共享风险基因网络。剔除游离节点(CYB5A、TSKS、ACACB 等),得到了包含2654个节点和4282 个互作对的最大子网。共享风险基因网络的网络直径为12,聚类系数是0.034,网络平均路径长度为4.972。
2.3 共享致病网络模块的挖掘和拓扑属性分析 共享风险基因网络被Newman 算法分解识别出17 个有统计学意义的关键功能模块。各模块的特征路径长度均小于6,无标度检验的指数参数为2~3,且连通度均符合幂律分布(KS 检验,P>0.05),反映所构建的网络符合无标度性质,见表1。

表1 各功能模块基本特征和拓扑属性
2.4 模块核心基因的识别 本研究识别了105 个核心基因,其中M3 模块中的核心基因数量最多,达到24 个。通过查阅GeneCards 和PudMed 数据库发现69 个基因已被证实与BC 或TC 相关,其中ESR1、RXRG、S100B 等25 个基因被证实与两种癌症均相关,见表2。

表2 各功能模块的核心基因
2.5 关键模块的功能富集分析 KEGG 富集分析显示,各模块主要富集于癌症通路、p53 信号通路、雌激素信号途径,见表3。进一步通过KEGG 网站查询挖掘各功能模块间的相互关系发现,17 个关键功能模块被划分为免疫系统(immune system)、癌症(cancer)、传染病(infectious disease)功能类别,见图2。
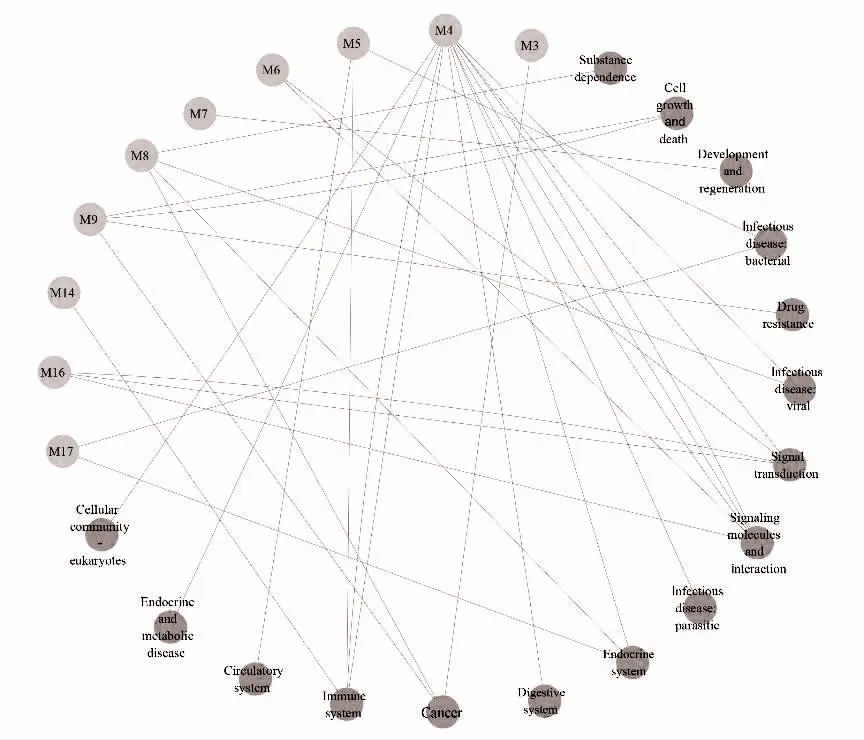
图2 功能模块与通路分类的关系

表3 各功能模块的KEGG 通路分析结果
2.6 生存分析 共得到16 个基因与BC 预后相关,5个与TC 预后相关。预后相关基因中有8 个未有文献报道与这两种疾病相关,见表4。

表4 各模块两种癌症预后相关的核心基因
3 讨论
生物学功能的实现并不是由单一基因实现的,往往需要通过多个基因的协同作用。在PPI 网络中,基因通过协同作用实现不同的生物学功能,表现为形成多个成簇聚合的高度紧密连接的节点,从而把复杂网络按照不同的生物学功能划分为不同的模块,又称为网络模块化。根据这一特征,可结合目前存在的多种网络分析方法[14]将复杂的生物分子网络分解为具有统计学意义的关键功能模块,从而深入的挖掘疾病的分子机制。
本研究把共享风险基因网络分解为17 个功能模块,各模块在直径、特征路径长度、连通度等拓扑参数接近,且拟合优度检验均符合无标度网络属性。各模块富集到的通路与两种癌症密切相关,如M17中的雌激素信号通路(hsa04915),其产物雌激素作为BC 的良好预测和预后的标记物,在肿瘤微环境中具有潜在的免疫调节作用[15]。在TC 中,雌激素促进了肿瘤细胞的增长并参与调节与TC 预后最为密切的血管生成和癌细胞转移[16]。M16 模块中的Hippo信号通路(hsa04390)是促进BC 细胞增殖和迁移以及肿瘤生长所必需途径[17]。在TC 中,多个致病因子(如SNHG15、ST6GAL2)的异常表达是通过作用于Hippo 信号通路来促进肿瘤的发生发展[18,19],反映出所识别出的每一个模块均与两种疾病的发病机制密切相关。对各模块富集到的KEGG 通路进行类别划分发现,M4 模块参与多个功能类别,该模块及其核心基因对于揭示两种疾病的共同发病机制具有最重要的价值。
此外,本研究所构建的网络中每一模块符合无标度性,即存在着少数连通度较高的核心基因,它们位于中枢位置,维系模块网络的稳定和模块内基因的相互调控作用,从而维持模块的生物学功能。当它们发生突变或者受到干扰时,模块对应的生物学功能也会发生异常,从而导致疾病的发生发展。由此,本研究对各模块节点进行泊松分布检验,识别出105 个核心基因。经GeneCards 和PudMed 检索发现,S100B、ESR1、CDKN2A 等基因被报道与BC 和TC 的癌细胞的生长、分化、侵袭和转移密切相关。如ESR1,其编码雌激素受体,还作为转录因子去调控DNA 结合、转录激活以及激素结合。作为BC 研究的焦点,ESR1 的激活突变是BC 治疗中获得性内分泌耐药的关键机制[20]。同时,ESR1 的高表达与侵袭性TC 行为密切相关,还可作为预测甲状腺外延伸、淋巴结转移、远处转移和高TNM分期的指标[21]。且ESR1 在本研究中的BC 和TC 疾病组中均呈高表达,反映出该基因的多效性。同时,未见DLG2、KCNJ2、GHR 等与BC 或TC 相关基因的报道,因此对其研究有助于挖掘两种癌发病关联的潜在机制。本研究BC 和TC 疾病组中均低表达的DLG2 是潜在的肿瘤抑制因子,其异常表达会导致多种癌症上皮细胞恶性增殖和赘生性转化[22,23]。此外,参与了细胞中钾通道的形成的KCNJ2,研究发现[24],KCNJ2 在侵袭性胃癌细胞中呈高表达水平,而沉默KCNJ2 会显著降低胃癌细胞的侵袭和转移能力。Liu H 等[25]在小细胞型肺癌的研究中发现,KCNJ2 高表达会促进癌细胞生长并使其对化学治疗药物不敏感,表明DLG2 在多种癌症中异常表达,表现出其基因多效性,可作为癌症新的预后标志物和治疗靶标。此外,本研究对各模块生存分析显示,模块M3 中的预后相关基因数量最多,且部分基因与两种癌症预后均相关,提示M3 模块对两种癌症的预后有着重要的研究价值;且其预后相关的基因中CD19、ACAN、RORB 等8 个基因未曾有文献报道,有望成为新的癌症预测靶点。
综上所述,本研究基于基因多效性理论对两种癌症的构建共享功能模块网络进行分析,挖掘它们致病的关键功能模块以及核心基因,为两种癌症发病关联机制以及预后提供了新的思路。
