不同负载方式Fe/改性MCM-41催化剂的制备及其降解亚甲基蓝性能研究
2021-11-14吴凤龙王岳俊
宋 瑾,吴凤龙,王岳俊
(1.河套学院生态治理与绿色发展院士专家工作站,内蒙古巴彦淖尔015000;2.河套学院生态与资源工程系)
目前,国内印染废水深度处理单元主要采用吸附处理、膜分离、高级氧化深度处理、高级生物处理等技术。其中,高级氧化深度处理技术中的传统Fenton化学氧化技术仍是常见的处理工序[1-3]。与均相类Fenton反应相比,非均相类Fenton反应因使用了固体催化剂便于分离和再利用,且催化剂所用载体可根据需求灵活选择和改性,在去除有机污染物方面有客观的现实意义和经济意义[4-6]。研究发现,在固体催化剂使用过程中,载体表面的物化性质和催化剂制备方法的不同均对活性组分的分散性和催化剂的性能产生影响[7]。因此,一方面,采用有机官能团修饰或功能化法对常见载体MCM-41分子筛进行改性,可以改变纯MCM-41表面的物理特性,进而影响催化性能。另一方面,采用浸渍负载活性金属、原位负载活性金属和NaBH4还原负载活性金属方式所制备的3类催化剂,负载方式的不同也会影响催化性能。但是,关于两者协同作用影响非均相类Fenton催化剂性能的研究较少。
鉴于此,以自制二元共聚物聚[苯乙烯—3-(甲基丙烯酰氧)丙基三甲氧基硅烷]制备改性MCM-41,并以其为载体,分别采用浸渍负载活性金属方式、原位负载活性金属方式和NaBH4还原负载活性金属方式制备铁基催化剂,同时以亚甲基蓝溶液模拟有机废水进行催化氧化研究。采用XRD、TEM、FTIR、XPS、压汞测试等手段分析了改性分子筛孔结构及活性组分Fe与改性MCM-41的相互作用对催化性能的影响并给出协同作用机理,为找出影响非均相类Fenton催化剂性能的因素提供理论借鉴。
1 实验部分
1.1 试剂与仪器
实验试剂:苯乙烯、3-(甲基丙烯酰氧)丙基三甲氧基硅烷、过硫酸铵,均为分析纯;十六烷基三甲基溴化铵、正硅酸乙酯、碳酸氢钠、无水乙醇、氢氧化钠、七水硫酸亚铁、亚甲基蓝、硼氢化钠、MCM-41,均为分析纯。
实验仪器:PHS-3E型pH计;DGG-9070AD型电热恒温鼓风干燥箱;OLB-100C型恒温摇床;LPHHS型数显恒温水浴锅;JJ-1型精密增力电动磁力搅拌器;SH-D(Ⅲ)型循环水式多用真空泵;YZ1515x型蠕动泵;JP-080S型超声波清洗机;QSH-VTF-1400T型真空管式炉;T9S型紫外可见分光光度计;D8 ADVANCE型多晶(粉末)X射线衍射仪;Tecnai G2 20型透射电子显微镜;FTIR-7600型红外光谱仪;Thermo ESCALAB 250XI型X射线光电子能谱;AutoPore IV 9510型高性能全自动压汞仪。
1.2 催化剂合成方法
将200 mL蒸馏水、10 mL苯乙烯、1.13 mL 3-(甲基丙烯酰氧)丙基三甲氧基硅烷、7.5 mL 20 mg/mL碳酸氢钠、0.7 mL无水乙醇加入到装有搅拌器和冷凝管的三口烧瓶中,并置于75℃的水浴锅内。搅拌10 min后滴加12 mL质量浓度为10 mg/mL的过硫酸铵,滴速为0.4 mL/min。恒温6 h后过滤,得到白色乳液。取乳液75 mL,依次将1.5 g十六烷基三甲基溴化铵、0.75 g氢氧化钠加入到装有搅拌器和冷凝管的三口烧瓶中,于65℃的水浴锅内搅拌3 h。滴加9.4 mL正硅酸乙酯,滴速为0.94 mL/min,恒温30 min后置于反应釜内,于120℃的烘箱中晶化12 h。过滤,用蒸馏水清洗滤饼至滤液pH等于7,80℃下烘干滤饼,研磨后得到改性MCM-41原粉。将原粉平铺在瓷舟内于管式炉内焙烧,升/降温速率为1℃/min,100、200、300、400℃各恒温1 h,500℃恒温3 h。研磨后得到改性MCM-41。
称取0.2 g改性MCM-41,加入0.11 g七水硫酸亚铁和15 mL蒸馏水,室温搅拌24 h后以转速为6 000 r/min的离心机离心3 min,弃去上清液,80℃下烘干得到Fe/改性MCM-41催化剂A。
称取0.2 g改性MCM-41,加入0.11 g七水硫酸亚铁、15 mL蒸馏水、0.004 5 g硼氢化钠,室温搅拌24 h后以转速为6 000 r/min的离心机离心3 min,弃去上清液,80℃下烘干得到Fe/改性MCM-41催化剂C。
Fe/改性MCM-41催化剂B的制备过程与上述制备方式相似,不同处是在反应釜内加入0.44 g七水硫酸亚铁共同晶化。
1.3 测试与表征
1)FTIR测试。采用KBr压片法制备样品,使用FTIR-7600型红外光谱仪,分辨率为4 cm-1,扫描次数32次。
2)XRD测试。采用D8 ADVANCE型多晶(粉末)X射线衍射仪,设定Cu靶,工作电压为40 kV,工作电流为100 mA,步宽为0.01°,扫描速度为0.5(°)/min,扫描角度为5~90°(2θ)。
3)TEM测试。采用Tecnai G2 20型透射电子显微镜拍摄形貌及面扫描。
4)XPS测试。采用压片法制样,AlKa射线为激发源,C 1s结合能为285 eV进行荷电校正,功率为150W,尺寸为500 μm,通过能量为50 eV,能量步长为0.05 eV。
5)压汞测试。采用AutoPore IV 9510型高性能全自动压汞仪,在真空条件下将汞注入样品管中,进行低压测定后将其放入高压站进行分析。
2 结果与讨论
2.1 XRD分析
为了明确改性MCM-41和催化剂A、B、C的结构信息,特进行XRD表征,结果见图1和图2。由图1可知,纯MCM-41在2θ为2.3°处出现较明显的d(100)衍射峰,4.0°和4.6°处出现较弱的d(110)和d(200)衍射峰,这是二维六方结构的特征峰。改性MCM-41出现了较弱的d(100)衍射峰,且向高角度方向移动,而d(110)和d(200)两处的衍射峰消失,表明虽然改性MCM-41结晶性下降、有序性降低,但分子筛骨架仍然存在(见2.4节FTIR分析)。出现这种现象的原因是在改性分子筛制备过程中,单元聚苯乙烯在焙烧阶段燃烧后留下了孔洞,导致分子筛孔结构发生改变所致。文献[8]报道,2θ为22.5°处是MCM-41中SiO2无定型衍射峰。由图2可知,催化剂A、B、C在该处的衍射峰均有所偏移,但偏移程度不同,其中催化剂A和C向高角度偏移较明显,这可能是活性组分Fe与载体改性MCM-41的相互作用或者载体的孔结构发生改变导致。此外,2θ为25.9、30.1、35.6°为Fe2O3特 征 峰,2θ为18.4、35.6、42.8°为Fe3O4特 征峰,表明催化剂A、B中活性组分Fe以Fe2O3和Fe3O4的形式存在。但是,与催化剂A、B相比,催化剂C衍射谱图中没有寻找出任何含铁元素物质的衍射峰,表明该催化剂中的Fe为无定形态或者粒径较小[9],这种现象在硼氢化钠还原过渡金属时易出现。另外,催化剂的颜色A(土黄色)、B(棕黄色)、C(红棕色)是Fe在催化剂制备过程中被空气氧化造成的,而颜色的差别也进一步说明了由于制备方法的差别而使Fe以不同的价态与粒径存在催化剂中。

图1 改性MCM-41和MCM-41的XRD谱图Fig.1 XRD patterns of modified MCM-41 and MCM-41
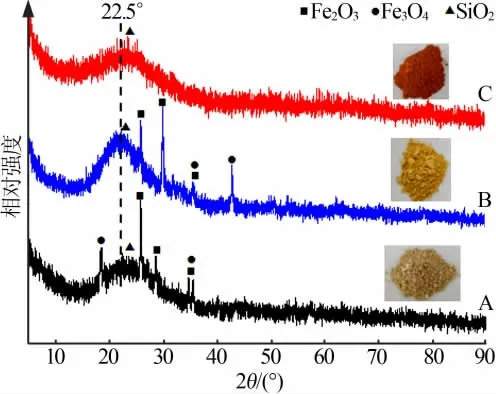
图2 不同负载方式制备的Fe/改性MCM-41的XRD谱图Fig.2 XRD patterns of Fe/modified MCM-41 catalysts with different loading methods
2.2 TEM分析
进一步探究不同负载方式制备的铁基催化剂表面形貌及元素分布,将催化剂A、B、C进行TEM表征,结果如图3a~c所示。3种催化剂表面均有粒径约200 nm左右的孔洞出现,且催化剂A的孔洞与催化剂B、C相比更多更均匀。均匀的孔道更有利于Fe2+的分散和负载,对提高催化剂活性是有利的。
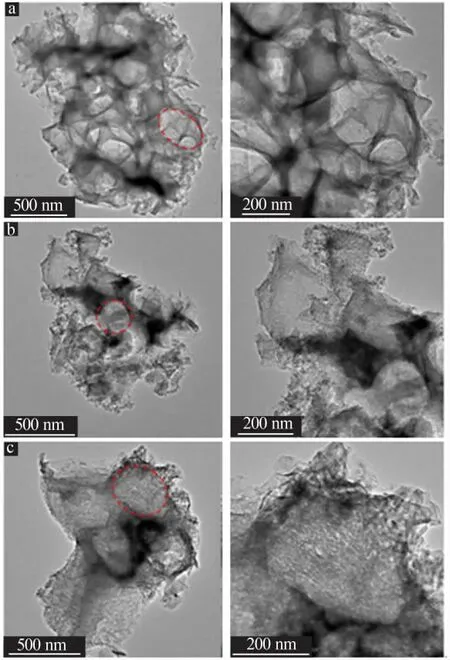
图3 Fe/改性MCM-41催化剂A(a)、B(b)和C(c)的TEM照片Fig.3 TEM images of Fe/modified MCM-41 catalyst A(a),B(b)and C(c)
图4 a~c为3种催化剂的元素分布(Mapping),铁均匀分散在改性MCM-41表面及孔道内。对于催化剂C而言,虽然XRD没有检测到铁元素的衍射峰,但是铁元素确实存在于催化剂中。

图4 Fe/改性MCM-41催化剂A(a)B(b)和C(c)的Mapping图Fig.4 Mapping images of Fe/modified MCM-41 catalyst A(a),B(b)and C(c)
2.3 压汞测试分析
由TEM可以看出催化剂为200 nm左右的大孔居多,这是由于改性焙烧过程形成的。为了进一步获取催化剂孔径分布、总孔体积、总孔表面积、孔隙度、实密度、表观密度等的物理特性,特进行压汞测试。图5a~b和表1分别为催化剂A、B、C的累积孔容曲线、孔径分布曲线和孔结构参数。由图5a可知,催化剂A、B、C的孔容分别为1.47、1.94、2.78 mL/g。从图5b看出,A催化剂的最可几孔径为1 652 nm,孔径主要集中分布在10 nm~4 μm。B催化剂的最可几孔径为57、438、1 905 nm,孔径主要集中分布在10 nm~4 μm。C催化剂的最可几孔径为31 nm和153 nm,孔径主要集中分布在10 nm~1 μm;而最可几孔径为28 031 nm,孔径主要集中分布在5~70 μm的大部分为颗粒之间的堆积孔,可能是硼氢化钠还原Fe2+时还原产物Fe粒径较小,较小的Fe易聚集堆积而出现较多的堆积孔[10]。很明显,3种负载型催化剂均以大孔为主,且含有少量的中孔。由表1可以看出,催化剂A的实密度大于催化剂B和C,表明单位体积内催化剂A的活性组分较多。此外,催化剂A的总孔隙面积、孔隙度、平均孔径均小于催化剂B和C,这进一步说明了孔结构的不同也是影响催化剂性能差异的因素。
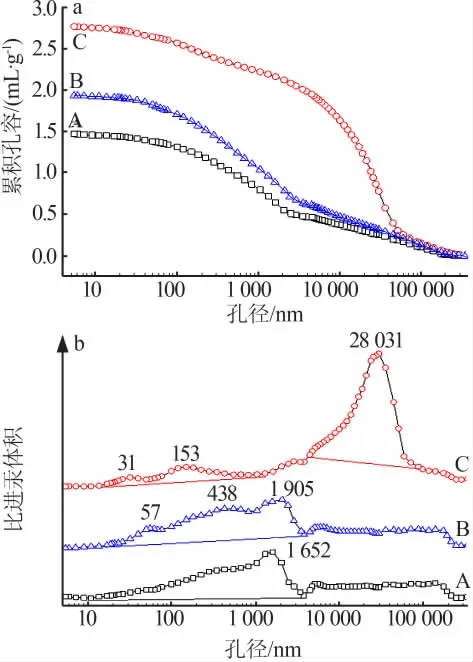
图5 不同负载方式制备的Fe/改性MCM-41的累积孔容曲线图(a)和孔径分布曲线(b)Fig.5 Cumulative pore volume curves and pore size distribution curves of Fe/modified MCM-41 prepared by different loading methods
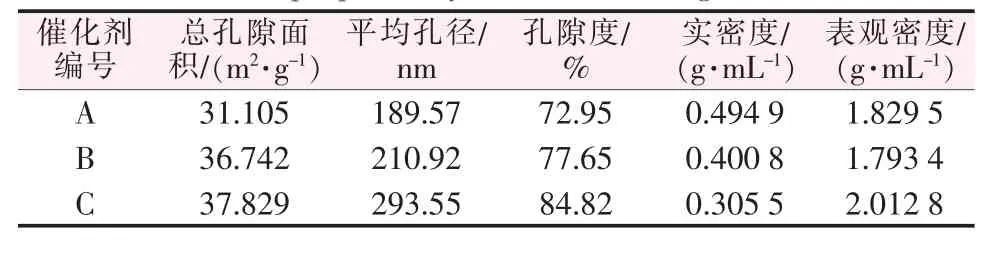
表1 不同负载方式制备的Fe/改性MCM-41的孔结构参数Table 1 Pore structure parameters of Fe/modified MCM-41 prepared by different loading methods
2.4 FTIR分析
将纯MCM-41和催化剂A、B、C进行红外表征,结果如图6所示。由图6可以看出,波数1 080 cm-1为O—Si的不对称伸缩振动峰,波数786 cm-1为O—Si的对称伸缩振动峰,为MCM-41特征骨架,说明不同负载方式制备的催化剂都保持了MCM-41的骨架结构,Fe元素的引入并没有影响改性MCM-41的晶格结构。
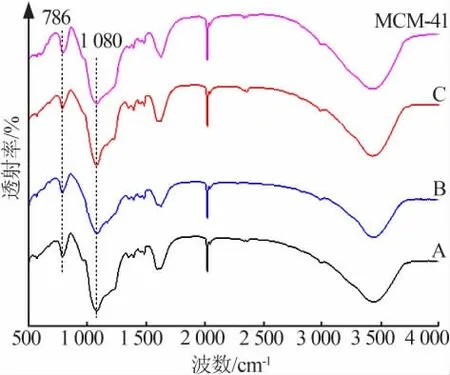
图6 不同负载方式制备的Fe/改性MCM-41催化剂、MCM-41的红外光谱图Fig.6 IR spectra of Fe/modified MCM-41 catalysts with different loading methods and MCM-41
2.5 XPS分析
为了分析负载Fe元素的价态及Fe与改性MCM-41的相互作用,特进行XPS表征,结果如图7~9所示。由3种催化剂的全谱图(图7a)可以看出,结合能在711.7 eV左右和725.3 eV左右的谱峰分别归属为Fe 2p3/2和Fe 2p1/2,结合能在535 eV处左右的谱峰归属为O 1s,表明催化剂表面存在Fe和O两种元素。由3种催化剂的Fe 2p高分辨图谱(图7b)可知,与文献报道的Fe 2p定位在711.0 eV和724.5 eV处的结合能相比[11-12],3种催化剂Fe 2p的结合能都向更高结合能的方向发生偏移,偏移的原因是Fe中的电子向分子筛转移,使两者之间产生了相互作用[13],这可能有利于吸附与降解亚甲基蓝分子[14-15]。由 图8和 图9中3种 催 化 剂 的Fe 2p和O 1s高分辨分峰谱图可以看出,峰位于711.3 eV和724.9 eV左右的两个谱峰均归属于Fe2+,峰位于713.4 eV和727.7 eV左右的两个谱峰均归属于Fe3+[11-12],说明制备的催化剂中含有Fe2+和Fe3+,这与XRD的测试结果是一致的。在O 1s的高分辨分峰图谱中,位于530.3 eV的谱峰归属于Fe—O,位于531.2 eV的谱峰归属于Si—OH,位于532.8 eV的谱峰归属于吸附水[11]。值得注意的是,Si—OH谱峰的峰形没有明显变化,表明Fe并没有大量进入改性MCM-41晶格中,这与FTIR的分析结果是一致的。在改性MCM-41晶格中是否存在少量Fe,还待进一步考察。虽然XRD没有检测到催化剂C中铁元素的衍射峰,但是通过XPS分析,铁元素确实存在于催化剂C中,这也与Mapping分析相互印证。综上所述,Fe与改性MCM-41间相互作用也是导致催化性能出现差异的原因。

图7 不同负载方式制备Fe/改性MCM-41的XPS谱图(a)和Fe 2p谱图(b)Fig.7 XPS spectra(a)and high resolution spectra(b)of Fe 2p in Fe/modified MCM-41 prepared by different loading methods
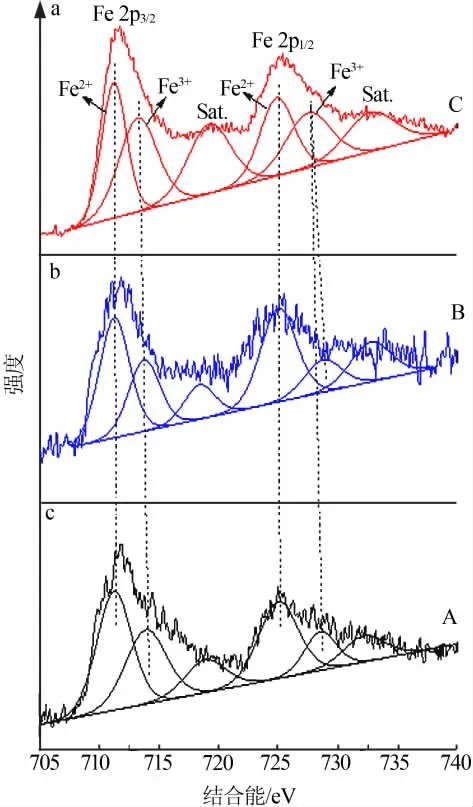
图8 不同负载方式制备Fe/改性MCM-41的Fe 2p高分辨分峰谱图Fig.8 High resolution peak separation spectra of Fe 2p in Fe/modified MCM-41 prepared by different loading methods
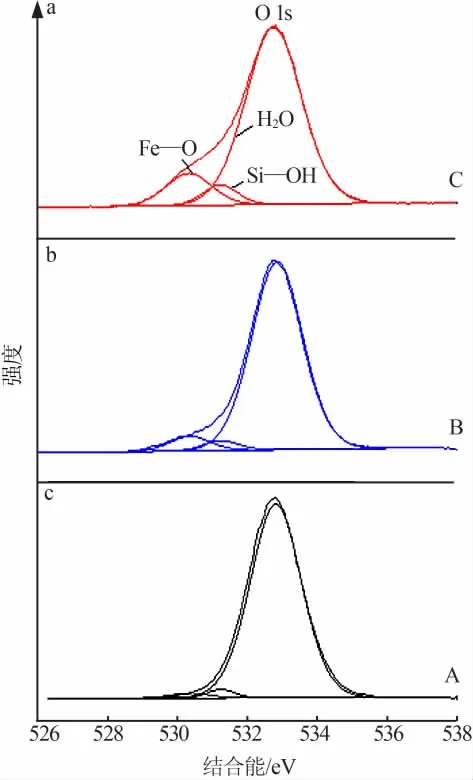
图9 不同负载方式制备Fe/改性MCM-41的O 1s高分辨分峰谱图Fig.9 High resolution peak separation spectra of O1s in Fe/modified MCM-41 prepared by different loading methods
2.6 亚甲基蓝去除率的测定
图10 为亚甲基蓝吸收光谱扫描曲线图(a)和亚甲基蓝浓度与吸光度标准曲线(b)。测定过程:将0.1 g催化剂、100 mL质量浓度为100 mg/L亚甲基蓝溶液置于250 mL的锥形瓶内,超声分散3 min后用1∶1的盐酸溶液调节pH为3~4,移入10 mL 30%的H2O2后将其置于150 r/min的恒温摇床内反应一定时间。用转速为10 000 r/min的离心机离心1 min,取上清液在其最大吸收波长处测其吸光度并根据标准曲线方程计算去除率。
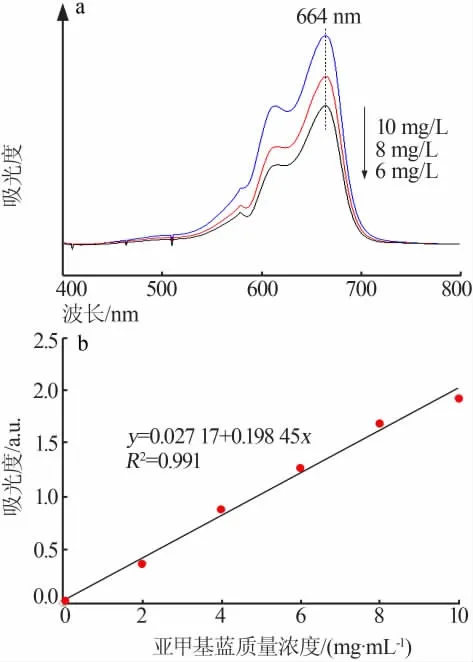
图10 亚甲基蓝吸收光谱扫描曲线(a)和亚甲基蓝浓度与吸光度标准曲线(b)Fig.10 The scan chart of methylene blue(a)and the standard curve of concentration and absorbance of methylene blue(b)
2.7 不同负载方式Fe/改性MCM-41催化剂对亚甲基蓝降解性能的影响
按2.6节的步骤分别以催化剂A、B、C催化H2O2氧化亚甲基蓝,结果如图11所示。由图11可以看出,不同催化剂具有不同的催化性能,催化性能由强到弱的顺序为A、B、C。即通过浸渍负载活性金属方式制备的催化剂性能最佳,25 min时亚甲基蓝去除率可达81.2%;通过原位负载活性金属方式制备的催化剂性能居中,25 min时亚甲基蓝去除率为73.5%;而通过NaBH4还原负载活性金属方式制备的催化剂性能最差,25 min时亚甲基蓝去除率仅为19%。因此,在同种载体、相同降解时间的条件下,不同活性组分的负载方式对催化剂的性能是有影响的。结合2.1~2.5节表征分析,这归因于:1)活性组分与载体的相互作用不同;2)催化剂的制备方式不同,导致孔结构发生改变。
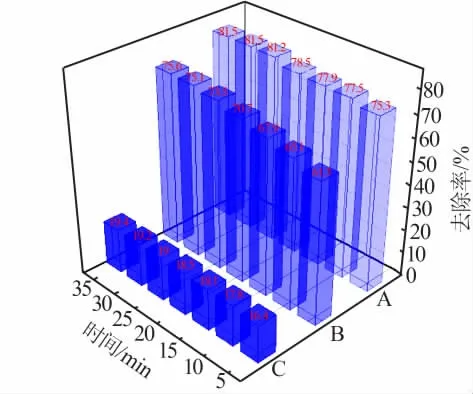
图11 不同负载方式Fe/改性MCM-41催化剂对亚甲基蓝去除率的影响Fig.11 Effect of Fe/modified MCM-41 catalysts with different loading methods on removal rate of methylene blue
2.8 协同作用机理分析
结合2.1~2.7节的表征分析及性能测试,可以推测出以浸渍负载活性金属方式、原位负载活性金属方式和NaBH4还原负载活性金属方式所制备催化剂性能不同的原因是:Fe—改性MCM-41间相互作用与催化剂孔结构两者的协同。一方面,在合成改性MCM-41过程中,由于二元共聚物聚[苯乙烯-3-(甲基丙烯酰氧)丙基三甲氧基硅烷]中单元苯乙烯被烧掉后形成孔洞,导致改性分子筛的孔结构发生改变。另一方面,活性组分Fe与改性MCM-41的相互作用改变了负载金属铁的化学环境。两方面的协同作用使不同负载方法制备催化剂的性能产生差异。
3 结论
1)通过浸渍负载活性金属方式制备的催化剂25 min时亚甲基蓝去除率为81.2%;通过原位负载活性金属方式制备的催化剂25 min时亚甲基蓝去除率为73.5%;通过NaBH4还原负载活性金属方式制备的催化剂25 min时亚甲基蓝去除率为19%。
2)XRD、TEM、FTIR、XPS、压汞测试等手段分析了不同负载方式制备的Fe/改性MCM-4催化剂性能出现差异的原因:焙烧后,聚[苯乙烯-3-(甲基丙烯酰氧)丙基三甲氧基硅烷]中单元苯乙烯使改性分子筛形成孔洞,改变了改性分子筛的孔结构;活性组分Fe与改性MCM-41的相互作用改变了负载金属铁的化学环境。上述两者的协同作用使催化剂的性能各异。
