富Li铝电解质体系分子动力学模拟
2021-11-06徐宏雷
韩 硕,徐宏雷,甄 鹏
(神华准能资源综合开发有限公司,内蒙古 鄂尔多斯 010300)
铝是仅次于钢铁的第二大金属,熔盐铝电解槽是其工业生产的核心装备。近年来,我国铝电解工业发展迅猛,原铝产量和消费量均连续多年超过全球总量的50%[1],在大型铝电解槽与特大型铝电解槽的设计开发与工程技术方面取得了举世瞩目的成就。
同时,国产Al2O3原料(特别是河南与山西等地所产Al2O3)普遍富含Li、K等碱金属杂质[2],因Li、K等碱金属比Al的电负性更低,随着Al2O3原料的不断添加与消耗,Li、K等杂质元素将在电解质熔体中持续累积。采用富Li/K国产Al2O3原料的电解槽运行一定时期后,电解质熔体中一般含有较高的Li/K含量,有些(LiF+KF)含量甚至高达10 wt.%,使得电解质初晶温度和溶解氧化铝能力明显降低,导致铝电解槽工艺控制、能量平衡与物料平衡愈加困难。采用富Li/K国产Al2O3原料的铝电解槽普遍存在电流效率偏低、能耗指标和效应系数偏高等突出问题,这已成为我国铝电解工业可持续发展中亟待解决的行业难题。因此,对于LiF-NaF-AlF3-Al2O3体系的结构与性质的研究势在必行,只有深入了解富锂盐电解质体系的结构与性质才能设计出合理的电解质体系。
由于铝电解质的高温强腐蚀特性,其微观结构的研究中常规实验手段难以进行,需要理论计算的方法进行辅助[3]。尽管富锂盐低温电解质体系的实验研究已经取得了诸多进展,目前实验研究主要集中在体系的基本物理化学性质上,如熔盐体系的初晶温度[4-5]、表面张力、密度[6]、粘度和电导率[7]等,然而,对于体系的微观离子结构的认知较少。
以往在对体系的微观结构进行探究时,主要采用动力学模拟方法,如:经典分子动力学模拟(IPMD)和第一性原理分子动力学模拟(FPMD)。IPMD又称作原子间势分子动力学模拟,是一种基于力场(等同于势函数)的一种模拟方法,其原理就是将体系中每种原子之间的作用力用函数的形式来描述,这种函数就被成为势函数[8-9]。利用此方法模拟过程的精度受多方面因素的影响,其中最重要的就是势函数本身的精确性,这个精确性指的是势函数本身能否模拟或者重现体系中原子间的相互作用,而势函数在计算体系性质时又严重依赖于势函数中相关势参数的准确性。因此,在利用IPMD对体系的离子结构进行模拟计算时,必须事先构建模拟体系的势函数以及相关的势参数,这无疑限制了经典分子动力学模拟的应用范围。为了克服经典分子动力学模拟的局限性,可以把第一性原理与分子动力学模拟结合起来,实现第一性原理分子动力学模拟。Car和Parrinello等人[10]于1985年提出了目前最常用的第一性原理分子动力学模拟方法,被称作CPMD或FPMD模拟方法。FPMD模拟方法采取如下方法直接计算体系中原子核间的相互作用及其位置演化,具体包括:① 利用第一性原理计算模拟体系在一固定时间间隔的电子结构与能量。② 计算体系中各原子核在该时间间隔的受力。③ 利用经典力学计算各原子核下一时间间隔的位置坐标。④ 重复上述过程,得到体系在相空间的运动轨迹。基于以上计算过程,FPMD模拟避开了经典分子模拟所需要的势函数以及势参数,提高了计算的精度。
因此,本文采用IPMD和FPMD模拟相结合的方法来研究LiF-NaF-AlF3-Al2O3熔盐体系的离子结构和输运性质,采用IPMD模拟LiF-NaF-AlF3-Al2O3熔盐体系的初始化状态,再采用FPMD模拟过程用以修正IPMD的计算结果,从而使模拟计算的结果和实际值趋于一致。另外,IPMD和FPMD相结合的计算方法和数据分析过程也为今后微观离子体系的研究拓宽了道路。
1 计算方法和参数设置
1.1 IPMD模拟参数设置
IPMD模拟的LiF-NaF-AlF3-Al2O3低温电解质体系化学组成(CRt(([LiF]+[NaF])/[AlF3])=1.3)如表1所示。初始的结构模型由Packmol软件[11]随机的将离子投放到指定大小的盒子中得到,模拟盒子的密度设置为1.90~2.01 g/cm3[12]。IPMD模拟采用Buckingham势函数,相关的势参数如表2所示。在IPMD模拟中,使用Verlet蛙跳算法求解牛顿运动方程,时间步长设置为0.1 fs。Ewald求和算法被用于处理体系中粒子间的库仑相互作用和偶极相互作用,同时Buffer宽度设置为0.5 Å,能量计算精度为10-5kcal/mol。短程相互作用的截断半径为15 Å,离子的电荷设置为Li (+0.67e),Na (+0.67e),Al (+2.01e),F (-0.67e)和O (-1.34e)。模拟盒子系统首先在NPT系综下以1.01 MPa压力被升温到1200 K,以保证LiF-NaF-AlF3-Al2O3体系处于熔融状态,模拟时长500 ps。之后使用NVT系综继续进行1 ns的结构驰豫,所有的IPMD计算均在Forcite模块中完成。
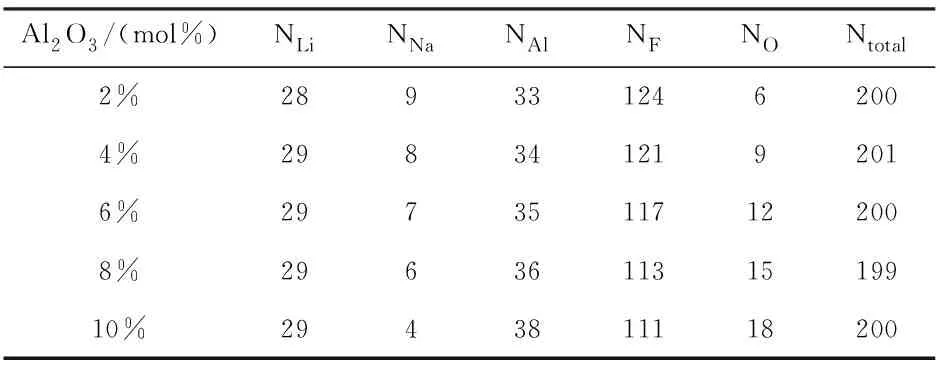
表1 LiF-NaF-AlF3-Al2O3体系的组成成分
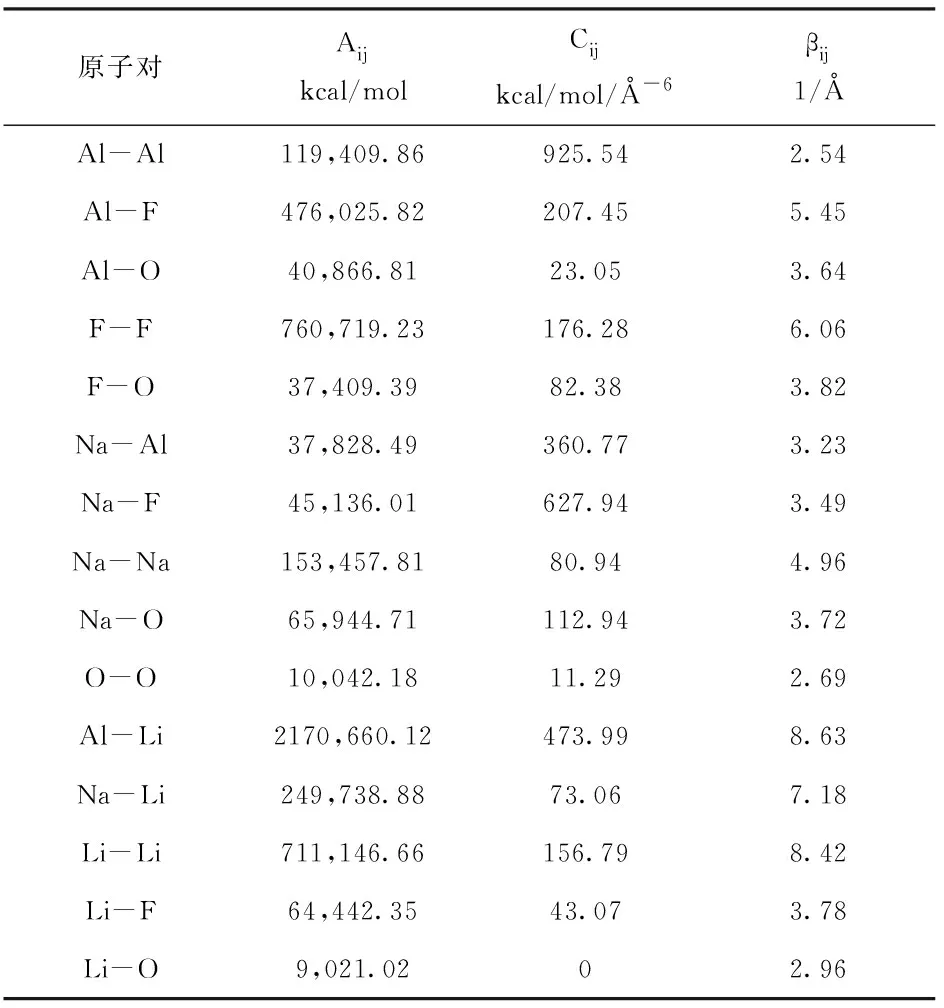
表2 LiF-NaF-AlF3-Al2O3熔盐电解质体系的Buckigham势参数拟合结果
1.2 FPMD模拟参数设置
IPMD模拟得到的LiF-NaF-AlF3-Al2O3体系的稳定构型被用于FPMD模拟,FPMD模拟采用广义梯度近似(GGA)中的Perdew-Burke-Ernzerhof(PBE)相互交联函数。使用Ultrasoft超软赝势来处理离子实和价电子的相互作用,其中,Na2s22p63s1电子,Al3s23p1电子,Li2s1电子,F2s22p5电子和O2s22p4电子被视为价电子。将DFT-D2方法应用于色散校正中提高仿真精度。FPMD计算采用520 eV的截断能和2×2×2 k-point网格。周期性边界条件被用于模拟盒子中来消除边界效应的影响,使得到的模拟结果更加合理。FPMD模拟采用固定粒子数,体积和温度的NVT系综,温度设置为1200 K。为了得到更加精确的熔盐离子结构和输运性质,熔盐体系先在NVT系综下进行5000步模拟,得到接近真实体系的熔盐离子结构,然后再进行10,000步的结构弛豫,模拟时间步长设置为1 fs,总的模拟时间为15 ps。最后得到的10,000帧粒子轨迹信息用于统计分析LiF-NaF-AlF3-Al2O3熔盐体系的离子结构和输运性质,FPMD模拟均在CASTEP模块中进行。
2 结果与讨论
2.1 LiF-NaF-AlF3-Al2O3熔盐体系微观构型
FPMD模拟得到的LiF-NaF-AlF3-Al2O3熔盐体系的稳定构型如图1所示,各配体构型是根据径向分布函数(RDF)中的截断半径定义得到的。从图1可以看出Na离子随机分布在模拟盒子中,并且离子之间的距离较大,这是因为Na-Na离子对的极性差异较小,且无共价特性。不同于之前的富钾盐电解质,LiF-NaF-AlF3-Al2O3体系中Li离子较为活跃,熔盐中Li离子并不是单独存在,而是像Al离子一样与F离子和O离子形成络合离子,这一点在之前的Li-F和Li-O离子对势能面计算中也有所体现。但是整体上看,熔盐中主要络合离子类型仍然以Al-F配离子为主。LiF-NaF-AlF3-Al2O3熔盐体系虽然失去了长程有序状态,但是局域离子结构仍然保持着短程有序状态。
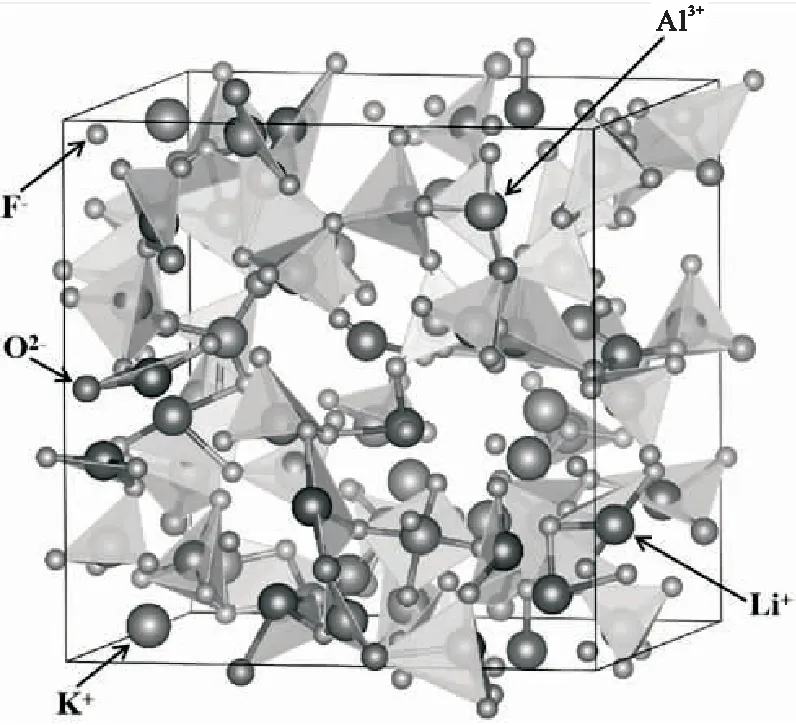
图1 LiF-NaF-AlF3-Al2O3熔盐体系的稳定构型(Al2O3/mol%=6)
2.2 不同离子对的径向分布函数
LiF-NaF-AlF3-Al2O3熔盐体系中不同离子对的径向分布函数(RDF)如图2所示。在r> 10 Å之后,不同离子对的RDF数值逐渐趋近于1,这与熔体近程有序,远程无序的结构形式吻合。通过观察图2a可知,Al-O和Al-F离子对的RDF的第一峰值高而尖(1.71 Å附近,一般RDF的第一峰值半径大小代表着某一离子对的平均键长),意味着熔盐中O和F离子与Al离子的结合能力较强,容易形成较为复杂的Al-O-F络合离子,并且Li-O和Li-F的RDF曲线也存在明显的峰值。一般来说,Al-Al离子对的RDF曲线可以反应LiF-NaF-AlF3-Al2O3熔盐体系的聚合程度,因为F和O离子可能会作为桥离子连接两个Al离子形成更为复杂的空间构型。此观点也在图2b中的Al-Al离子对的RDF曲线上得到了证实,曲线中存在两个较为明显的峰值,分别位于3.33 Å和4.73 Å处。3.33 Å处的峰值大约是Al-O(F)离子对的第一峰值半径1.71 Å的两倍左右,对应着熔盐中的Al-O(F)-Al构型,即O(F)离子作为桥离子连接两个Al离子。第二峰值出现在4.73 Å左右对应的是更为复杂的Al-O(F)-Li-O(F)-Al构型,这说明Li离子也参与了熔盐中离子的络合。

图2 LiF-NaF-AlF3-Al2O3不同离子对的RDF曲线 (Al2O3/mol%=6)
2.3 Al-F和Li-F离子对的平均配位数
LiF-NaF-AlF3-Al2O3熔盐体系中Al-F和Li-F络合离子的平均配位数(CN)可以通过对RDF曲线积分得到,CN积分曲线如图3所示(Al2O3mol%=6)。

图3 Al-F和Li-F离子对的平均配位数积分曲线(Al2O3/mol%=6)
当截断半径分别取2.45 Å和2.87 Å时,Al-F和Li-F络合离子的平均配位数分别为4.75和3.95,同理不同Al2O3浓度的Al-F和Li-F络合离子的CN也可以积分得到,如表3所示。随着Al2O3浓度逐渐增大,Al-F络合离子的平均配位数也逐渐增大,这是因为O离子的引入,代替了部分与Li离子配位的F离子,自由F离子重新被Al离子捕获,从而增大了Al-F络合离子的配位数。Li-F络合离子的平均配位数在3~4之间,并且随着Al2O3浓度逐渐增大而逐渐减小。LiF-NaF-AlF3-Al2O3熔盐体系中存在Li-F络合离子,与富钾盐电解质不同,这也是富锂盐电解质的氧化铝溶解度较低的原因所在。

表3 不同氧化铝浓度下LiF-NaF-AlF3-Al2O3体系中Al-F和Li-F配离子的平均配位数计算结果
2.4 LiF-NaF-AlF3-Al2O3熔盐体系的输运性质
LiF-NaF-AlF3-Al2O3熔盐体系的输运性能对铝电解过程至关重要,根据FPMD模拟得到的轨迹信息可以统计得到离子自扩散系数如图4所示,可以看出LiF-NaF-AlF3-Al2O3熔盐中离子的扩散能力为Li>Na>O>F>Al。Li离子的扩散能力强于Na离子,这是因为Li离子的半径较小,在熔体中的扩散更容易。而熔盐中的Al、F和O离子的扩散能力较弱,是因为Al离子与F离子和O离子之间的极性相差较大,易在熔盐体系中形成较为复杂的络合离子,从而降低了其扩散能力。随着Al2O3浓度的增大,熔盐中不同离子的扩散系数均有所降低,这是因为桥F和桥O离子的存在,熔盐中形成了体积更为庞大的Al-F-Al和Al-O-Al络合离子,使得离子的扩散变得困难。

图4 LiF-NaF-AlF3-Al2O3熔盐中离子的自扩散系数
LiF-NaF-AlF3-Al2O3熔盐体系的粘度和离子电导率如图5所示。熔盐的离子电导率的计算结果在2.81~2.98 S/cm之间,并且随着Al2O3浓度的增大,熔盐的离子电导率逐渐减小,这是因为Al2O3的引入使得熔体的结构变得复杂,离子的扩散能力降低,电导率也有所降低。离子电导率计算结果远高于常规的富钾盐电解质体系1.2~1.5 S/cm,这与实际规律相符[13]。此外,离子电导率的计算结果2.81~2.98 S/cm与文献报道的nLiF-2.2NaF-AlF3体系 2.85~3 S/cm相接近[12,14],且略低于该体系,这是因为氧化铝的加入导致的。粘度的计算结果在1.84~2.23 mPa·s之间,并且随着Al2O3浓度的增大,熔盐的粘度逐渐升高。
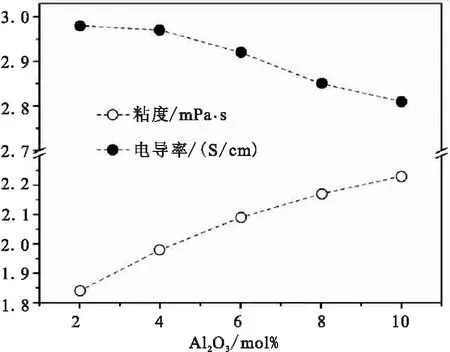
图5 LiF-NaF-AlF3-Al2O3熔盐体系的粘度和电导率
3 结 论
采用IPMD和FPMD相结合的方法研究了LiF-NaF-AlF3-Al2O3熔盐体系的微观离子结构与输运性质。得到主要的结论如下:
(1)在LiF-NaF-AlF3-Al2O3熔盐体系中,不仅存在着简单的Al-F络合离子,同时由于桥O和桥F离子的存在,这些络合离子集团会结合在一起形成体积更为庞大的复杂离子构型。
(2)Al-F络合离子的平均配位数在4~5之间,Li离子在熔盐中会与F离子结合形成络合离子,从而与Al离子产生竞争,降低Al-F络合离子的配位数。Li-F络合离子的平均配位数在3~4之间,并且随着Al2O3浓度的增大而逐渐减小。
(3)LiF-NaF-AlF3-Al2O3熔盐中离子的扩散能力为Li>Na>O>F>Al,随着Al2O3浓度的增大,熔盐中不同离子的扩散系数均有所降低。熔盐的离子电导率的计算结果在2.81~2.98 S/cm之间,粘度的计算结果在1.84~2.23 mPa·s之间。
