妇炎净片中氯化两面针碱含量测定方法改进
2021-11-06廖玉群康志华付向红杨雷熊家伟
廖玉群,康志华,付向红,杨雷,熊家伟
江西药都仁和制药有限公司,江西 樟树 331200
妇炎净片处方由当归、鸡血藤、苦玄参、地胆草、菥蓂、五指毛桃、横经席、两面针、柿叶九味组成,具有清热祛湿,调经止带之功效,用于湿热蕴结所致的带下病、月经不调、痛经;慢性盆腔炎、附件炎、子宫内膜炎等证。方中两面针具有祛风通络,渗湿止痛,抗炎抗菌等活性[1]。本品为江西药都仁和制药有限公司拟进行二次开发的重点产品之一,现执行国家标准YBZ13472005-2009Z,标准中已有苦玄参、两面针、地胆草、鸡血藤的薄层鉴别,以及氯化两面针碱的含量测定,其中氯化两面针碱的含量规定每片中不得少于0.08 mg;而市场上秦皇岛皇威制药有限公司的同品种氯化两面针碱的含量规定每片中不得少于0.22 mg(执行国家药品标准YBZA12312006)。故在二次开发中,为进一步提高产品质量及质量的可控性,对本品中氯化两面针碱的含量及其测定方法进行了研究,对原标准含量测定方法进行了改进,改进后的方法操作简便,重复性良好,结果准确,具有专属性;同时提高了氯化两面针碱的内控含量指标,由原标准中每片不少于0.08 mg,提高为每片不少于0.20 mg。
1 仪器与试药
1.1 仪器
高效液相色谱仪:岛津LC-20AD、安捷仑1260 Ⅱ;电子天平:CP224S 赛多利斯(d=0.1 mg)及CP225D(d=0.01 mg、0.1 mg)。
1.2 试药
氯化两面针碱对照品(中国食品药品检定研究院,批号:110848-201802,含量以91.0%计,使用前不需干燥处理);妇炎净片样品(由江西药都仁和制药有限责任公司提供,批号:191001、191002、191003、200301、200302、200303、200801、200802、200901、210301);
乙腈为色谱纯(购自美国天地公司);水为超纯水;其他试剂均为分析纯。
2 方法与结果
2.1 原注册标准方法
2.1.1 色谱条件与系统适用性试验 以十八烷基硅烷键合硅胶为填充剂;以乙腈-水-三乙胺(23∶77∶0.3)(用磷酸调pH 值为3.0)为流动相;检测波长为328 nm。理论板数按氯化两面针碱峰计算应不低于2 000。
2.1.2 对照品溶液的制备 精密称取氯化两面针碱对照品适量,加甲醇制成每1 mL 含5 µg 的溶液,摇匀,即得。
2.1.3 供试品溶液的制备 取重量差异项下的样品,研细,取约2.0 g,精密称定,精密加入1%盐酸甲醇溶液50 mL,稳定重量,加热回流1 h,放冷,再称定重量,用1%盐酸甲醇溶液补足重量,滤过,精密吸取续滤液10 mL,过中性氧化铝柱(100-200目,10 g,内径1.5 cm),用甲醇50 mL 洗脱,收集洗脱液,蒸干,残渣加甲醇使溶解,转移至25 mL量瓶中,加甲醇稀释至刻度,摇匀,即得。
2.1.4 测定法 分别精密吸取对照品溶液与供试品溶液各20 µL,注入液相色谱仪,测定,即得。
在大量的生产实践中发现,本法重现性不理想,影响因素较多,结果偏差较大,加样回收率偏低,不能真实反映出氯化两面针碱的含量。
2.2 改进后的方法
2.2.1 色谱条件与系统适用性试验[2-7]以十八烷基硅烷键合硅胶为填充剂;以乙腈:0.02 mol/L KH2PO4(22∶78);检测波长为328 nm。理论板数按氯化两面针碱峰计算应不低于2 000。
2.2.2 对照品溶液的制备 精密称取氯化两面针碱对照品适量,加甲醇制成每1 mL 含10 µg 的溶液,摇匀,即得。
2.2.3 供试品溶液的制备[8-9]取重量差异项下的样品,研细,取约1.0 g,精密称定,精密加入1%盐酸甲醇溶液50 mL,稳定重量,加热回流1 h,放冷,再称定重量,用1%盐酸甲醇溶液补足重量,滤过,精密吸取续滤液25 mL,蒸干,残渣加水20 mL 使溶解,用稀盐酸调pH 1~2 后,用三氯甲烷振摇提取3 次,每次20 mL,合并三氯甲烷液,蒸干,残渣用甲醇使溶解,定量转移至25 mL 量瓶中,加甲醇稀释至刻度,摇匀,即得。
2.2.4 测定法 分别精密吸取对照品溶液与供试品溶液各20 µL,注入液相色谱仪,测定,即得。
2.2.5 分析方法确认 按照《中国药典》2020 年版四部通则9101 药品质量标准分析方法验证指导原则,选取线性关系、专属性、定量限、回收率、精密度、耐用性、稳定性作为确认指标,对改进后的分析方法进行了确认,结果如下。
线性关系考察:以氯化两面针碱的浓度(µg/mL)为横坐标X,峰面积为纵坐标Y,绘制标准曲线,得回归方程Y=46.869X-2.162,相关系数r=0.999 9(n=6)。氯化两面针碱质量浓度在2.06~51.50 µg/ mL范围内与峰面积线性良好。
专属性试验:色谱图中,氯化两面针碱能够较好地分离,保留时间适中,阴性无干扰,专属性好。
精密度试验:取氯化两面针碱对照品溶液(浓度为20.60 µg/ mL),连续进样6 次,测定峰面积,结果的RSD 为0.66%(n=6),小于2%,表明仪器精密度良好。
稳定性试验:按供试品溶液制备方法制成供试品溶液(批号191001),同时取氯化两面针碱对照品溶液(20.60 µg/ mL),分别在放置0、2、4、6、8、10、12、24 h 进样,测定峰面积,结果的RSD 为1.3%(n=8),表明溶液在24 h 内稳定性良好。
重复性试验:取样品(批号191001)6 份,按正文中含量测定项下方法试验,测定氯化两面针碱的含量,结果6 份样品所测得氯化两面针碱平均含量为289.22 µg/片,RSD 为1.6%(n=6),表明方法重复性良好。
定量限:当信噪比为10∶1 时,氯化两面针碱的浓度为20 ng/mL。

图1 高效液相色谱图:A.阴性样品;B.供试品;C.对照品
2.3 原注册标准方法及改进后的方法对比
取样品(批号191001),分别采用原注册标准方法及改进后的方法,进行精密度、加样回收率以及耐用性试验,测定氯化两面针碱的含量,检测结果汇总见表1~3。

表1 精密度试验结果对比(µg/片)

表2 加样回收率试验结果对比(%)

表3 耐用性试验结果对比(µg/片)
结果,原注册标准方法精密度、加样回收试验及耐用性试验的RSD 分别为1.9%、3.3%和1.9%,改进后方法的精密度、加样回收试验及耐用性试验的RSD 分别为0.4%、0.7%和0.3%,表明改进后的方法较原方法的精密度、准确度及重现性更理想,原方法加样回收率偏低,改进后的方法平均加样回收率为99.5%,表明结果准确,更能真实地反应出本品中氯化两面针碱的含量,可用于本品的质量控制。
3 样品测定
取本品,分别按原方法及改进后的方法测定氯化两面针碱的含量,改进后的方法检测的氯化两面针碱的含量均比原方法检测结果高;考虑到不同产地的两面针中氯化两面针碱的含量有高有低[10],拟将氯化两面针碱的含量由每片不得少于0.08 mg,提高至每片不得少于0.20 mg。检测结果见表4。
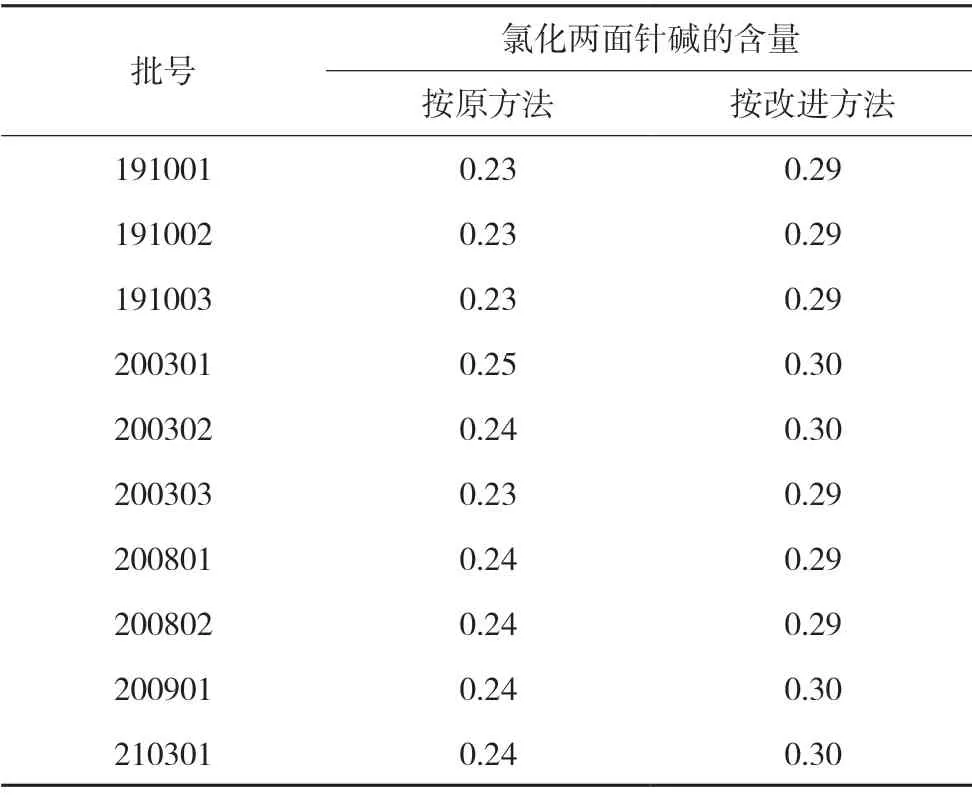
表4 10批样品中氯化两面针碱的含量(μg/ 片)
4 讨论
4.1 流动相的选择
氯化两面针碱属苯骈菲喧类生物碱,其结构中碱性氮原子与固定相未键台酸性硅醇基的相互作用,使得色谱峰的展宽拖尾,峰漂移及峰对称性差,导致分离效能低。故对比了在流动相中加添加缓冲盐及有机胺改性剂的效果,结果乙腈-0.02 mol/L 磷酸二氢钾溶液(22∶78)的效果优于乙腈-水-三乙胺(23∶77∶0.3)(用磷酸调pH 值为3.0),况且三乙胺对色谱柱的损害不易逆转,故选择采用乙腈-0.02 mol/L 磷酸二氢钾溶液(22∶78)为流动相。
4.2 检测波长的选择
氯化两面针碱对照品溶液及供试品溶液在271 nm及328 nm 处均有吸收峰,在328 nm 下的色谱图中杂质干扰峰稍少,故仍选取328 nm 作为检测波长,不做修订。
4.3 供试品溶液的制备
修订的方法采用1%的盐酸酸化的甲醇进行回流提取,可以使游离的两面针碱结合成为无机酸盐,从而能够提高氯化两面针碱的提取率;但同时使得提出的杂质也增多;大多数游离生物碱都是亲脂性的,故甲醇液蒸干后,加水使溶解调pH1~2,再用三氯甲烷萃取,以达到分离纯化的目的。原方法中采用通过中性氧化铝柱进行分离纯化,结果,在去除杂质的同时,将目标成分氯化两面针碱同时损失了一部分,造成检测结果比真实含量偏低,结果不准确。
试验中,对各试验条件进行了优化筛选,如不同提取溶剂、回流提取时间、萃取次数、流动相比例等,结果以正文条件为佳。所建立的方法操作简便,专属性强,重复性理想,结果准确,优于原方法,能真实地反应本品中氯化两面针碱的含量。可用于妇炎净片的质量控制。
