内质网自噬—内质网质量控制新途径
2021-11-04伍美婷江俊麟
伍美婷,江俊麟
(中南大学湘雅药学院药理学系,湖南 长沙 410078)
内质网(endoplasmic reticulum,ER)是由生物膜构成的片状囊腔和小管状腔相互联通而成。ER是参与细胞内蛋白质合成、折叠,脂质代谢及钙储存等的重要细胞器。某些应激状态,如氧化应激、炎症、缺氧、钙代谢紊乱可导致蛋白质未折叠或错误折叠,干扰ER稳态,从而引发ER应激(endoplasmic reticulum stress,ERS)。在ERS早期,ER通过启动未折叠蛋白反应来减少蛋白质翻译、促进蛋白质折叠及ER相关降解,缓解ER压力。当细胞发生持续的ERS时,未折叠蛋白反应将导致细胞功能障碍,诱导细胞凋亡。自噬是指受损的细胞器或蛋白质通过溶酶体途径降解,从而确保细胞内环境稳定的过程。自噬最初被认为是一个非选择性的过程。新的研究发现自噬过程也具有选择性,其通过受体蛋白靶向清除细胞内受损的细胞器及蛋白聚合物。作为一种新发现的选择性自噬,ER自噬被报道在传染性疾病、癌症等病变中发挥重要作用。
1 内质网应激
片状ER主要分布在核周,向内与外核膜连接;管状ER通常形成网络状结构,分布于整个细胞中。根据形态,ER可分为粗面ER和滑面ER。粗面ER表面附着大量核糖体,是蛋白合成与加工的场所;滑面ER表面无核糖体附着,具有解毒、合成脂质、糖原及调节细胞内钙离子稳态的功能[1]。多种病理性刺激均可引发细胞内环境紊乱,降低ER内蛋白质折叠效率,并导致未折叠蛋白和错误折叠蛋白在ER内大量聚集,最终引起ERS。
ERS主要包括3种反应形式:未折叠蛋白反应(unfold protein response,UPR)、ER超负荷反应(endoplasmic reticulum-overload response,EOR)和固醇调节级联反应,前两者是由蛋白质错误折叠导致,后者是ER中胆固醇损耗所致。
UPR是研究最为广泛的信号通路之一,早期ERS通过激活UPR,维持ER稳态并恢复细胞功能。UPR通过调节下游感受器蛋白肌醇需求因子(inositol-requiring enzyme,IRE1)、活化转录因子6(activating transcription factor 6,ATF6)、类蛋白激酶ER激酶(protein kinase R-like ER kinase,PERK)启动适应性反应,降低mRNA浓度,减少蛋白质翻译,诱导泛素-蛋白酶体依赖的ER相关降解(ER-associated degeneration,ERAD)及提升分子伴侣(GRP78、GRP94)表达,参与折叠和稳定ER腔内蛋白,恢复ER稳态,最终缓解细胞压力[2]。当ERS持续高水平存在,UPR将会导致细胞功能障碍,诱导细胞凋亡。
EOR是指正确折叠的蛋白质在ER上过度聚集,激活核因子κB(nuclear factor kappa-B,NF-κB)所致。研究显示,ER膜蛋白聚集损伤了Ca2+-ATP酶的功能并提高脂质双分子层对Ca2+的通透性,从而导致大量Ca2+从ER释放,诱导大量活性氧(reactive oxygen species,ROS)产生,后者可诱导NF-κB活化。UPR也可导致EOR。正常情况下,NF-κB与NF-κB抑制蛋白I-κB相互结合形成复合物位于细胞质,抑制NF-κB易位和活化。UPR下游信号中的IRE1α通路可通过降解I-κB,引起NF-κB核易位并使其激活;另一条PERK通路则通过减少I-κB翻译而激活NF-κB[3]。
固醇调节级联反应是由胆固醇缺乏引起。当发生ERS时,ER合成的胆固醇耗竭,激活ER膜上的固醇调节元件结合蛋白(sterol regulatory element binding protein,SREBP)和SREBP裂解激活蛋白(SREBP cleavage activating protein,SCAP),使其形成复合物并由ER转移至高尔基体。在高尔基体中SREBP被酶解为活性因子进入细胞核,激活与脂质合成有关基因的转录,维持细胞内脂质含量[4]。ERS时间过长或强烈活化可引起级联反应,导致细胞凋亡。
2 内质网自噬
自噬是细胞的重要保护机制,是通过消除不必要的蛋白质或受损细胞器来维持细胞稳态的分解代谢过程,常在营养缺乏或某些应激下发生。在真核细胞中,自噬分为大自噬、微自噬和分子伴侣介导的自噬。既往研究认为,自噬通过降解细胞内组分来维持细胞的能量代谢和存活,是一个高度保守的非选择性过程。新的研究表明,在酵母和真核生物细胞中存在选择性自噬,目的在于降解某些特定底物,如受损的细胞器、蛋白聚集物和入侵的病原微生物等。根据细胞器的不同,可将选择性自噬分为ER自噬、线粒体自噬、过氧化物酶体自噬及核糖体自噬等。2006年,Bernales等[5]首次在酵母中发现,持续的ERS和UPR可引起针对ER的选择性自噬,形成只含ER的自噬小体,并将其命名为ER自噬(ER-phagy)。
2.1 内质网自噬的产生ER-phagy是一条新的ER质量控制途径。正常情况下ER-phagy保持在一个较低水平以维持ER稳态。当机体受到刺激,如营养匮乏、病原体感染时,蛋白酶体途径无法降解ER内大量堆积的蛋白质,ER-phagy则作为一个“补偿”机制来恢复ER功能[6]。根据作用方式的不同,ER-phagy可分为内质网巨自噬(macro-ER-phagy)、内质网微自噬(micro-ER-phagy)和小泡吞饮内质网自噬(vesicular delivery)。macro-ER-phagy大致可分为4个步骤:①ER上的自噬受体通过其LC3相互作用结构域/Atg8相互作用结构域/GABARAP相互作用结构域与自噬双层膜相互作用,形成碎片并从ER上脱离;②自噬双层膜延伸将ER片段包裹成新的自噬小体;③自噬小体与溶酶体融合;④自噬小体被溶酶体酶降解(Fig 1)。ER微自噬是指溶酶体直接通过内吞的形式,将ER包裹进入溶酶体腔内降解的过程。小泡吞饮介导的ER自噬是指未折叠蛋白聚集物形成小泡从ER上脱落,进一步被溶酶体捕获并降解。目前,ER巨自噬是报道最为广泛的一类ER-phagy。
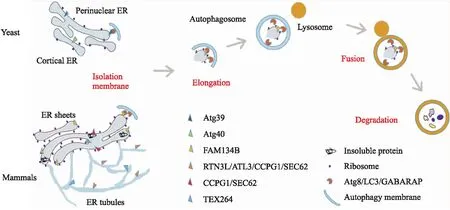
Fig 1 Macro-ER-phagy process
2.2 内质网自噬的作用作为一条新发现的ER质量控制途径,ER-phagy具有双重作用:一是在持续的ERS过程中,ER-phagy有利于隔离无法发挥正常功能的ER或是大量不能通过其他方式处理的错误折叠蛋白;二是在ERS减弱时,ER-phagy可减少膨胀的ER,使之形态恢复正常。
2.3 内质网自噬受体ER-phagy是由ER上的自噬受体介导的。研究发现,在酵母中存在2种ER-phagy受体,分别是Atg39和Atg40。在哺乳动物细胞ER膜上存在6种受体,分别是序列相似性家族134B(family with sequence similarity 134 member B,FAM134B)、浆膜蛋白3长剪切体(reticulon 3,RNT3L)、ATL3(atlastins 3)、分泌转运蛋白62(secretory translocation protein,SEC62)、细胞周期进程基因1(cell-cycle progression gene 1,CCPG1)和睾丸表达基因264(testis expressed gene 264,TEX264)(Fig 2)。ER-phagy受体主要有两个作用,一是识别需被降解的ER或ER腔内的未折叠蛋白,二是通过Atg8相互作用结构域(Atg8-interacting motif,AIM)或LC3相互作用结构域(LC3-interacting region,LIR)与自噬相关蛋白Atg8/LC3/GABARAP结合介导自噬体形成。

Fig 2 ER-phagy related receptors
2.3.1酵母内质网自噬受体 在酵母中,氮缺乏和雷帕霉素诱导的ER-phagy是由ER自噬受体Atg39和Atg40介导。作为Atg8结合蛋白,Atg39和Atg40均包含一个AIM,促进其与自噬膜上的Atg8结合。Atg39属单跨膜蛋白,定位于核周ER,主要参与核周ER的清除;Atg40包含一个与哺乳动物ER同源结构域(reticulon-homology domain,RHD)类似的结构,其对ER片段的脱落及ER膜的形态修复具有重要作用,主要参与皮层ER的清除[7]。由Atg39和Atg40介导的ER-phagy需Atg1、Atg8、 Atg11和Atg17的参与[7]。
2.3.2哺乳动物细胞内质网自噬受体
2.3.2.1 FAM134B FAM134B是首个发现的哺乳动物ER自噬受体,也是现今研究最为广泛的ER自噬受体。最初,FAM134B被认为是高尔基上的特定蛋白,主要参与感觉和自主神经病变的发病[8]。随后研究发现FAM134B包含一个RHD和LIR,其RHD能促进ER弯曲和片段形成,LIR能够锚定自噬泡上LC3/GABARAP,因此FAM134B被认为是一种ER自噬受体蛋白[9]。FAM134B可参与抵抗ERS,维持细胞内环境稳态。研究显示,在FAM134B敲低的U2OS细胞、FAM134B敲除的小鼠胚胎成纤维细胞或FAM134B下调的人胚肾293细胞,ER出现明显扩增,而过表达FAM134B可使ER片段增多并促进溶酶体降解[8]。除降解ER片段外,FAM134B还参与维持蛋白稳态。在人骨肉瘤细胞和小鼠胚胎成纤维细胞中,FAM134B可与ER膜上的钙连蛋白结合,识别错误折叠的原胶原后,启动ER-phagy将其清除[10]。
2.3.2.2 RTN3L RTN3是高度富集于管状ER中的一种弯曲膜蛋白。RTN3含有RTN3L和RTN3S 2种剪切体,其中RTN3L为ER-phagy受体。RTN3L的N末端包含6个LIR,能够与LC3/GABARAP结合。RTN3L以单体和多聚体形式存在。单体形式的RTN3L参与维持管状ER的形态;RTN3L多聚体则促进含RTN1、RTN4、REEP5和RTN3的管状ER弯曲变形,并通过LIR与LC3/GABARAP结合并包裹成自噬小体[11]。研究显示,氨基酸过度消耗,可激活RTN3L,启动ER-phagy。在RTN3-/-的小鼠胚胎成纤维细胞中,饥饿介导的相关管状ER蛋白的重塑受到影响。有趣的是,在RTN3-/-小鼠,ER并没有出现功能缺陷[12]。因此,RTN3是否参与ER-phagy调控有待进一步研究。
2.3.2.3 ATL3 ATL3是具有ER跨膜结构域的ER-phagy受体。与其他哺乳动物受体不同,ATL3的LIRs并不位于一个固有的无序区域,而是存在于一个胞质内的N端动态蛋白样GTPase结构域内。ATL3含有2个非经典的LIR即GABARAP相互作用模块(GABARAP interacting motifs,GIMs),其通过与GABARAP结合来促进ER-phagy,清除受损的管状ER[13]。ATL3通常以二聚化的形式来调节ER出口位点的丰度。研究证实,在哺乳动物ATLs家族蛋白中,ATL1、ATL2、和ATL3具有高度的同源序列。在ATL2敲除的HCT116细胞中,重新表达上述任何一个ATLs cDNA都可恢复饥饿诱导的ER-phagy。免疫共沉淀结果进一步显示,ATL2与ATL3可通过GTP酶结构域相互作用,提示ATL2可能与ATL3形成异源二聚体,介导ER-phagy[14]。此外,Chen等[15]研究发现,ATL3的Y192C和P338R突变可抑制ATL3与GABARAP结合,削弱ATL3介导的ER-phagy,并诱导I型遗传性感觉和自主神经病变(hereditary sensory and autonomic neuropathy type I,HSAN I)。
2.3.2.4 SEC62 SEC62是粗面ER转运复合物SEC61/SEC62/SEC63中的一个跨膜蛋白,主要参与富含UPR上调相关分子伴侣和折叠酶(如钙连蛋白、钙网蛋白、ERp72和 ERp57等)的ER片段清除。当UPR处于正常水平时,SEC63与LC3竞争性地结合SEC62,并与SEC61形成复合体,参与介导新合成的前体多肽转运至ER[16]。与FAM134B等自噬受体介导的ER-phagy不同,SEC62介导恢复性ER自噬,即ERS被解除后,SEC62通过其C端的LIR介导恢复性ER-phagy,恢复ER稳态。SEC62与癌症如肺腺癌、前列腺癌等的发生发展相关。在非小细胞肺癌、前列腺癌、甲状腺癌等癌细胞中,SEC62呈现高表达,进而增加其LIR与LC3/GABARAP结合的敏感性,促进受损ER清除,导致肿瘤对ERS耐受及肿瘤耐药性增加[17]。
2.3.2.5 CCPG1 CCPG1是非经典的ER自噬受体。CCPG1主要分布在核周ER,是唯一一个N端同时含有1个LIR和2个FIP200互作结构域(FIP200-interatcting regions,FIRs)的ER-phagy受体。FIRs与酵母ER自噬受体Atg39上的Atg11结合模块有较高的相似性,CCPG1可能通过其N端的FIR与FIP200作用结合,招募ULK1复合物,加强细胞对自噬信号的募集[18]。ERS发生时,激活的CCPG1通过C端与受损的ER及腔内聚集的蛋白连接,使CCPG1其从ER上脱离,促进其被新生成的自噬小泡包裹。在小鼠胰腺,由CCPG1介导的ER-phagy能够减少ER腔内蛋白聚集和UPR,维持胰腺腺泡细胞内蛋白稳态,而CCPG1缺失可导致ER中大量酶原蛋白堆积,促进细胞死亡,最终导致胰腺炎[19]。
2.3.2.6 TEX264 TEX264是含有单跨膜结构的ER-phagy受体蛋白,在其C末端含有1个LIR和由113个氨基酸构成的动态固有无序区域(intrinsically disordered region,IDR),后者环绕LIR,可避免LIR受空间位阻的影响,有利于LIR与自噬膜的连接。饥饿条件下,细胞内大部分ER-phagy是由TEX264介导[20]。相比于上述5个ER-phagy受体,TEX264在多个器官中呈高表达,且广泛分布于ER膜上。此外,TEX264与LC3/GABARAP家族蛋白结合效率更高且更牢固,其原因可能是由于TEX264主要表达于ER三口连接处,而自噬体膜正是在ER三口连接处形成。研究显示,在Hela细胞,分别敲除6种不同的ER-phagy受体(FAM134B、RNT3L、ATL3、SEC62、CCPG1和TEX264),当敲除TEX264时,对ER-phagy影响最明显,而同时敲除TEX264、FAM134B和CCPG1将进一步损害ER-phagy,提示TEX264可能是介导ER-phagy的主要受体[21]。
2.3.2.7 其他 除上述ER膜上的ER-phagy受体外,研究发现p62、胞质蛋白CALCOCO1及胞质C53也可能参与介导ER-phagy。在TCPOBOP处理的小鼠,p62可将LC3-Ⅱ阳性的自噬体招募到泛素-p62修饰的ER,启动ER-phagy,介导肝脏受损ER的清除。与野生型小鼠相比,敲除p62敲除后,小鼠肝脏ER出现膨胀和体积增大[22]。在饥饿诱导的ERS中,胞质蛋白CALCOCO1可作为可溶形ER-phagy自噬受体,其通过FFAT样结构域与管状ER蛋白VAPA/B作用,并通过其UIR或者LIR相互作用结构域与Atg8家族蛋白相互作用,介导管状ER-phagy发生[23]。在共翻译蛋白转运过程中核糖体发生停滞时,胞质蛋白C53可通过感受ER蛋白质毒性,与ER相关泛素折叠修饰连接酶UFL1及ER膜受体DDRGK1形成复合物,后者被ER上滞留的核糖体激活,并暴露C53上的非典型ATG8相互作用元件使其与ATG8相互作用招募自噬小体,导致特异性ER蛋白的降解[24]。
3 内质网自噬与疾病
ER-phagy是参与调控ER功能的主要方式之一,能有效清除细胞内受损的ER和错误的蛋白聚集体,维持细胞内环境稳定。研究显示,在一些传染性疾病、癌症、神经退行性疾病、糖尿病等疾病中,ER-phagy水平会发生改变,过度激活或抑制ER-phagy可导致细胞内受损ER和错误蛋白聚集体清除障碍,使机体病变加剧。
在病毒和细菌的感染周期中,ER-phagy被认为是宿主细胞的重要防御机制,能够清除ER中的细菌或病毒。Chiremel等[25]发现,ER-phagy可抑制埃博拉病毒在小鼠胚胎成纤维细胞中的复制,而敲除FAM134B后病毒的复制能力明显增强,表现为VP40蛋白和核蛋白表达的增加。另一ER-phagy受体RTN3通过与丙型肝炎病毒的非结构蛋白NS4B的AH2结构域竞争性结合,抑制AH2自身寡聚化,抑制病毒复制[26]。病毒也可通过某些机制逃避宿主的ER-phagy。例如,丙型肝炎病毒蛋白酶NS3可通过破坏FAM134B的RHD结构域,削弱FAM134B介导的ER-phagy[27]。提示ER-phagy可能成为治疗传染性疾病的新靶点。
近期研究发现,ER-phagy受体FAM134B、SEC62参与多种癌症的发生发展。在食管鳞状细胞癌和肝细胞癌中,FAM134B作为促癌因子,通过激活Akt来促进细胞增殖和迁移;而在结肠癌和乳腺癌中,FAM134B则发挥抑癌作用[28]。SEC62在多种癌细胞如前列腺癌、甲状腺癌细胞中呈高表达,通过启动ER-phagy缓解ERS引起的细胞损伤,进而促进癌细胞生长[17]。
C型尼曼皮克病是一种神经退行性病变,主要是由编码NPC1的基因(1061位点异亮氨酸-苏氨酸)突变引起,该突变可影响跨膜糖蛋白折叠,导致细胞内未酯化胆固醇堆积。研究发现,低表达FAM134B可增加突变型NPC1蛋白含量,而过表达FAM134B可启动ER-phagy,减少突变型NPC1蛋白聚集,抑制细胞内未酯化胆固醇堆积[29],提示干预ER-phagy可能成为治疗C型尼曼皮克病的新方法。
胰岛素基因突变所导致的青少年糖尿病(mutant INS-gene-induced diabetes of youth,MIDY)是一种Akita胰岛素原基因突变型糖尿病,主要是由Akita胰岛素原基因突变致使胰岛素原不能正确折叠,阻止正常胰岛素原加工为成熟胰岛素所引起。Cunningham等[30]发现敲除RTN3可增加突变型胰岛素原聚集物的堆积,而过表达RTN3可通过激活ER-phagy,清除突变型胰岛素原聚集物,恢复ER功能,促进胰岛素产生,提示靶向ER-phagy可能成为治疗MIDY的有效途径。
4 小结
到目前为止,已发现多种不同类型的ER自噬受体,即Atg39、Atg40、FAM134B、RTN3L、ATL3、SEC62、CCPG1和TEX264,进一步丰富了人们对ER-phagy的认识。ER-phagy是一把双刃剑,适度的ER-phagy可降解错误/未折叠的蛋白质及受损的ER,维持细胞稳态;而过度激活的ER-phagy又能引起细胞死亡。因此,针对不同疾病模型,以ER-phagy受体为疾病治疗靶点进行深入研究,阐明其调控过度激活/抑制的ER-phagy的具体机制,也有望为相关疾病的治疗提供新思路。
