利用固体NMR研究一种萜类有机分子在聚乙烯中的吸附行为
2021-11-03宋建会刘宣伯姚雪容张韬毅张龙贵
宋建会,刘宣伯,姚雪容,李 娟,张韬毅,张龙贵
(中国石化 北京化工研究院,北京 100013)
聚乙烯(PE)是一种应用广泛的高分子材料,作为包装材料具有价格便宜、力学性能优异、加工方式多样、透明度和柔软度好等特点,在农业、食品、药品等领域具有广泛的应用[1-2]。包装材料的阻隔性能需满足一定的要求,而氧气、水蒸气、挥发性化合物等小分子在膜中的吸附和迁移都会影响包装产品的货架寿命[3]。前期已有关于PE吸附醛、甲基酮、甲酯和含硫化合物等气味分子的研究报道,影响吸附的因素包括分子尺寸、极性、溶解性、气味分子的浓度以及聚合物的性质(如形貌、结晶度和极性等)[4-5]。本课题组曾报道了萜类有机分子在不同聚烯烃材质中的渗透行为,提出萜类有机分子的扩散系数和渗透系数与聚合物基体的结晶度和玻璃化转变温度有关[6]。目前对PE包装材料吸附的研究主要关注在吸附量及吸附速率方面,而有机分子吸附对PE基体的结构影响以及扩散机理等尚缺乏深入认识。
固体NMR技术是研究固态高分子材料结构和分子动力学的一种非常重要和有效的手段[7-8],在PE的相结构研究方面具有独特的优势,可同时对PE结晶相和非晶相的结构及分子运动进行研究[9-11]。
本工作采用固体NMR技术考察了PE吸附一种萜类气味有机分子前后的相结构及分子运动变化情况,以探索有机分子在PE中的吸附机理。
1 实验部分
1.1 试剂
PE:密度0.924 g/cm3,熔体流动指数(10 min)1.9 g(190 ℃,2.16 kg),中海壳牌石油化工有限公司;C10H18O:萜类气味有机分子,室温下为固体,易挥发,闪点65 ℃,密度0.992 g/cm3,在25 ℃下的饱和蒸气压为3.12 Pa,云南林缘香料有限公司。
1.2 吸附实验
采用Carver公司Auto4533型实验室热压机和厚度为0.5 mm的压片模具制备PE薄片。将压好的薄片置于装有C10H18O的密封容器中进行常温吸附,至薄片恒重后用于NMR测试。吸附饱和后的PE中C10H18O的含量约为0.2%(w),吸附后的试样记为PE/OM。试样粉碎后装入核磁转子(氧化锆,Kel-F帽子)中压实,测试过程中由于密闭充实的环境,且转子对小分子不吸附,可忽略吸附的C10H18O的脱附。
1.3 固体NMR测试
固体NMR测试在Agilent公司600 MHz Premium Compact+型核磁共振波谱仪上进行,1H和13C的共振频率分别为599.89,150.86 MHz,室温下测试。4 mm探头,转速8 kHz,1H和13C的90°脉宽均为4 μs。以金刚烷为标样优化90°脉冲及Hartmann-Hahn条件,且13C的化学位移(δ)以金刚烷的次甲基信号(δ=38.56)为参考定标。
13C CP/MAS(交叉极化、魔角旋转和高功率去偶)NMR实验的接触时间为1 ms,等待时间为4 s。13C SPE/MAS(单脉冲实验,魔角旋转和高功率去偶)NMR的循环等待时间为1 000 s。
13C自旋-晶格弛豫时间(T1)的测量采用Torchia脉冲序列,利用Origin的指数衰减公式对信号强度和间隔时间进行拟合得到PE各相的13CT1值。
2 结果与讨论
2.1 C10H18O对PE基材结构的影响
PE,C10H18O及 PE/OM 的13C CP/MAS NMR谱图见图1。从图1可看出,C10H18O的谱图较复杂,因此未对信号进行详细归属。对于PE,由于半晶聚合物的相组成按经典描述仅包含无定形相和结晶相,因此δ=32.9,31.1处的峰分别对应PE的正交晶相和非晶相[12]。PE/OM的谱图中只有PE的信号,这是因为即使PE基材吸附C10H18O至饱和后,PE/OM中的C10H18O含量依然很低,仅约为0.2%(w),因此很难观察到C10H18O的信号。吸附的C10H18O对PE的信号不造成明显干扰,因此吸附前后试样的NMR谱图差异可直接反映PE结构的变化。

图1 13C CP/MAS NMR谱图Fig.1 13C CP/MAS NMR spectra.
由于13C CP/MAS NMR谱图受交叉极化动力学的影响无法定量[13],可通过PE和PE/OM的13C SPE/MAS NMR谱图(即去偶13C NMR谱)(见图2)对结构进行定量研究,等待时间为1 000 s以确保碳核可以完全弛豫。
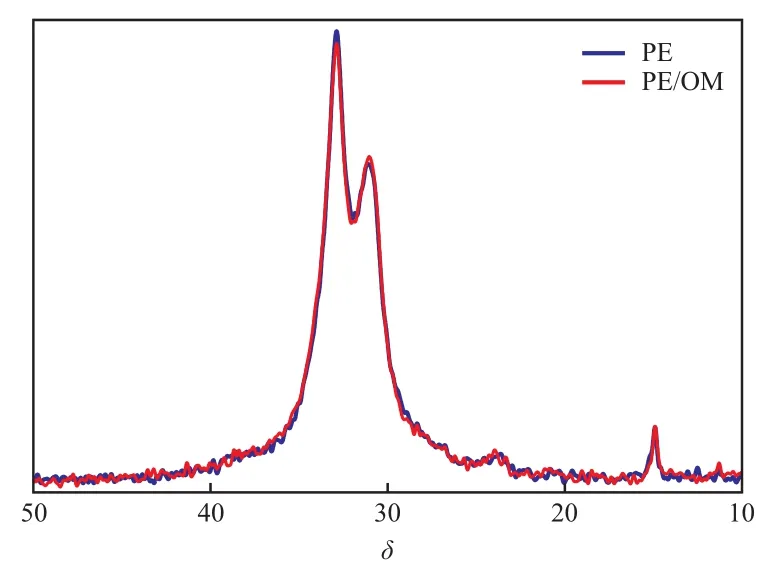
图2 PE和PE/OM的13C SPE/MAS NMR谱图Fig.2 13C SPE/MAS NMR spectra of PE and PE/OM.
从图2可以看出,PE和PE/OM的13C SPE/MAS NMR谱图存在一些差异,为了得到相区的含量需对谱图进一步进行分峰处理。PE作为半晶高分子,它的相结构除了结晶区和无定形区,在二者之间还存在界面区,界面区的分子链排列具有一定的有序程度,而且同时具有晶区和无定形区的结构特征[14-15]。利用dmfit软件对PE和PE/OM的13C SPE/MAS NMR谱进行分峰拟合,将谱图分成晶区、界面区和无定形区,分峰结果见图3。从图3可看出,该结果与Mattozzi等[16]报道的PE相结构一致:δ=33.1处的峰归属为晶区,δ=31.7处的峰归属为界面区,δ=31.2处的峰归属为无定形区,各相区的含量见表1。

图3 13C SPE/MAS NMR谱图的分峰拟合Fig.3 Peak fitting of 13C SPE/MAS NMR spectra.
从表1可看出,相比于PE,PE/OM晶区的含量基本不变,界面区的含量略有减少,无定形区的含量有所增加,这是由于吸附的C10H18O使PE的界面区部分发生“溶解”,形成更加无序的无定形区。
2.2 C10H18O对PE基材分子运动的影响
固体NMR还常用于研究体系的微观分子运动,是研究高分子体系中的分子运动非常有效的手段之一。通过测量13CT1研究PE基体在吸附C10H18O前后的分子运动变化。通常弛豫敏感的是分子运动的频率而非模式,体系中某一种弛豫行为常对应多种分子运动过程[17]。T1与体系中的高频运动相关,弛豫现象遵循Bloembergen-Purcell-Pound理论[18],对于PE,T1越大表明分子运动越慢。
图4为不同衰减时间时PE的13C NMR谱图。从图4可看出,信号强度随衰减时间的延长呈e指数函数衰减。晶区(δ=32.9)和无定形区(δ=31.1)信号强度的衰减趋势存在差异,晶区信号强度的衰减速率比无定形区慢得多,这是由于晶区和无定形区的链段运动性存在差异:晶区链段运动受到晶格限制,表现出刚性,因此具有较长的13CT1,表现为信号强度衰减慢;无定形区的链段运动剧烈,表现出柔性,因此具有较短的13CT1,表现为信号强度衰减快。

图4 Torchia法测量PE的13C T1Fig.4 13C NMR spectra of PE by Torchia pulse sequence.
对PE和PE/OM的13C T1结果用积分面积进行计算,积分范围为δ=26~42,积分面积随时间的衰减曲线见图5。从图5可看出,PE/OM衰减比PE快。PE的13CT1衰减曲线往往无法用单指数衰减公式拟合,因为实际数据包含不只一个组分,因此需要用多指数公式进行拟合[19]。在13C SPE/MAS NMR谱图分峰结果中,PE的相结构分为晶区、界面区和非晶区,相应地会有3种不同弛豫行为的分子运动。因此,将衰减曲线分成3个组分拟合13CT1:
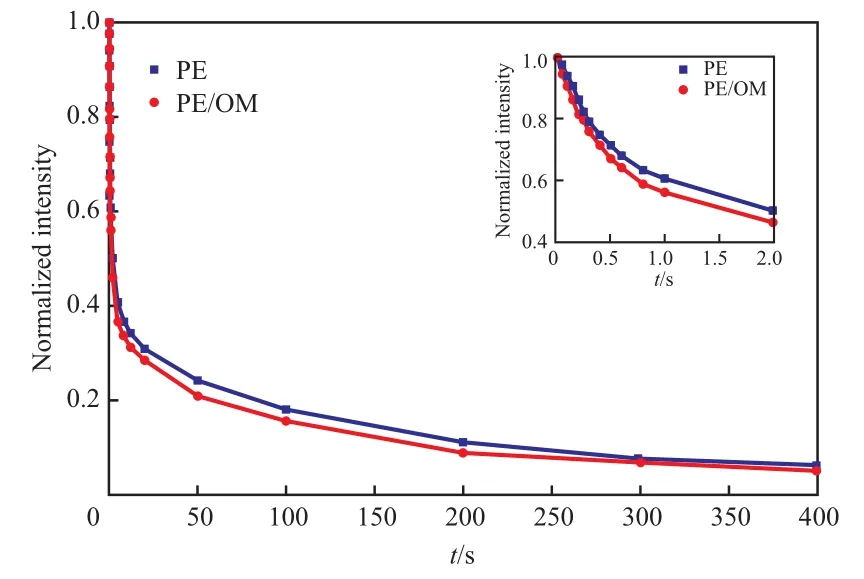
图5 PE和PE/OM的13C T1衰减曲线Fig.5 13C T1 decay curve of PE and PE/OM.

式中,t为延迟时间,s;Mtotal(t)为t时的总纵向磁化强度;Mi(0)(i=a,b,c)为各组分的初始磁化强度;T1i(i=a,b,c)为各组分的自旋-晶格弛豫时间,s。
PE和PE/OM的13CT1见表2。从表2可看出,由13CT1衰减曲线拟合得到的PE/OM三个组分的含量与PE相比变化不大,但三个组分的13CT1均比PE的短,这与图5中PE/OM曲线衰减更快相符。由于PE的结晶分子受晶格限制,运动能力弱,因此最大的13CT1对应PE的晶区;无定形区的分子链运动最快,因此最小的13CT1对应PE的无定形区;界面区的链段运动能力处于晶区和无定形区中间,因此中间的13CT1对应PE的界面区。此处拟合出的三个组分的含量与SPE/MAS NMR谱图分峰拟合的结果有所差异,这是因为两种实验方法及数据处理的原理不同:13C SPE/MAS NMR谱图分峰是根据晶区、界面区和无定形区的化学环境(化学位移)不同;而13CT1拟合则是根据不同的相区结构对应不同的弛豫行为,由13CT1衰减曲线拟合得到拟合度最高的三个相区含量。在相区比例变化不大的情况下,由13CT1衰减曲线拟合得到的含量仅有参考意义。吸附C10H18O后PE基材的界面区和无定形区的13CT1均变小,这主要是由于吸附的C10H18O的“溶剂化”作用使界面区和非晶区的的分子链运动变快。而吸附后晶区的13CT1也变小,表明溶剂还是一定程度地进入了晶区,使晶区的分子运动能力加快,但由于晶区的限制作用还不足以使晶区的含量发生变化。

表2 PE和PE/OM的13C T1值Table 2 13C T1 values of PE and PE/OM
3 结论
1)PE的相组成包含晶区、界面区和无定形区,吸附C10H18O至饱和后的PE/OM的晶区含量基本不变,界面区中有很小一部分转变为无定形区。
2)PE吸附C10H18O至饱和平衡后,晶区、界面区和无定形区的分子链运动均变快。
