PLA2G6基因突变致青年帕金森病的临床特征(附1例报告)
2021-11-02范成成蔡卫卫孙虹梅珊珊王慧许保磊马敬红许二赫陈彪
范成成,蔡卫卫,孙虹,梅珊珊,王慧,许保磊,马敬红,许二赫,陈彪
帕金森病(PD)是一种常见且复杂的神经变性病,典型的临床表现是运动迟缓、肌强直及静止性震颤。目前PD的病因及发病机制尚不明确。研究[1]表明,年龄、环境及遗传因素可能与PD的发病有关。其中,遗传因素在PD发病机制中的作用越来越受到关注,目前已克隆出二十余个PD相关的致病基因,其中PLA2G6被证明是与常染色体隐性遗传PD相关的致病基因,其突变可引起遗传性帕金森综合征14型(PARK14)[2]。
本文报道了一个可能致病的PLA2G6基因突变的新位点,并对此例PLA2G6复合杂合突变导致的青年PD(YOPD)患者的临床资料结合既往相关文献进行分析。
1 临床资料
患者,女,41岁,物流公司上班,无特殊接触史。主因“右下肢不自主抖动两年余,右下肢步态异常一年余”于2020年8月24日收入院。患者在两年多前(2018年7月)于劳累后出现右下肢抖动,静止时出现,紧张、生气后无明显加重,自觉长时间坐位或立位转变为平卧位时右下肢抖动明显,余肢体无抖动,无明显动作缓慢。2018年11月7日就诊于北京某医院,行多巴胺转运蛋白(DAT)-PET检查示左侧尾状核、壳核DAT分布略减低;D2-PET检查示左侧壳核多巴胺D2受体上调,结合DAT显像考虑PD可能。期间未服用抗PD药物,症状逐渐加重,近1年行走时出现右下肢拖步,右下肢出现卡顿感,有时需他人搀扶行走。偶有尿频、尿急,便秘多年,靠开塞露排便。无明显嗅觉障碍,无夜间睡眠中大喊大叫现象,无尿失禁。患者为独女,否认家族遗传病及类似病史。查体:卧位血压113/77 mmHg (1 mmHg=0.133 kPa),3 min立位血压120/80 mmHg。“关”期神经系统查体:神清语利,高级皮质功能粗测正常,双侧眼球活动正常,四肢肌力Ⅴ级,右侧上下肢肌张力增高(1分),左侧肢体肌张力正常。双上肢腱反射(),右下肢腱反射(),左下肢腱反射(+),双侧病理征(-),双侧轮替、对指、足拍打未见异常。双侧指鼻、跟-膝-胫试验稳准。闭目难立征(-),后拉试验(-)。行走时右上肢摆臂动作减少,右下肢足内翻,右下肢拖步。血、尿便常规、生化全项、甲状腺功能、同型半胱氨酸、铜蓝蛋白、传染病指标、凝血、风湿指标未见异常。脑部黑质超声:右侧黑质强回声(Ⅲ级)。头颅MRI未见小脑萎缩、脑异常铁沉积(图1)。EMG示双侧腓总神经运动传导远端潜伏期延长。MMSE(大学)30分,蒙特利尔认知评估量表(MoCA)30分,HAMD:1分,Hoehn-Yahr分期(“关”期未服药)1期。基因检查:全外显子基因检测发现该患者携带PLA2G6基因的复合杂合突变,其第7外显子检出c.991G>T(NM_003560)杂合突变,导致第331号氨基酸由天冬氨酸变为酪氨酸(p.D331Y),第12外显子检出c.1630A>G(NM_003560)杂合突变,导致第544号氨基酸由甲硫氨酸变为缬氨酸(p.M544V),经一代测序家系验证前者来源于其母亲(图2),其父亲因脑出血去世未能获取样本,其父亲无临床表现。治疗经过:入院后第2 d行多巴反应试验(美多芭口服,187.5 mg),基线UPDRS Ⅲ评分9分,服药后症状明显改善,1 h改善率为60%,2 h改善率为40%。给予盐酸苯海索片(1 mg,3次/d)联合森福罗(0.25 mg,3次/d),症状无明显改善。停用盐酸苯海索片,给予美多芭(62.5 mg,1次/d)联合森福罗(0.25 mg,3次/d),美多芭逐步加量至125 mg(2次/d),患者诉右下肢无力较前明显改善,右膝关节卡顿感基本消失。
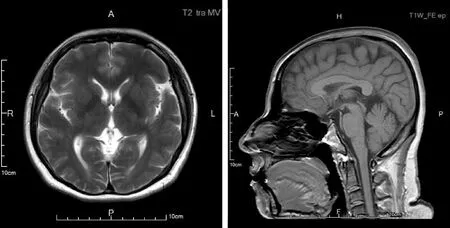
图1 头颅MRI未见小脑萎缩、脑异常铁沉积
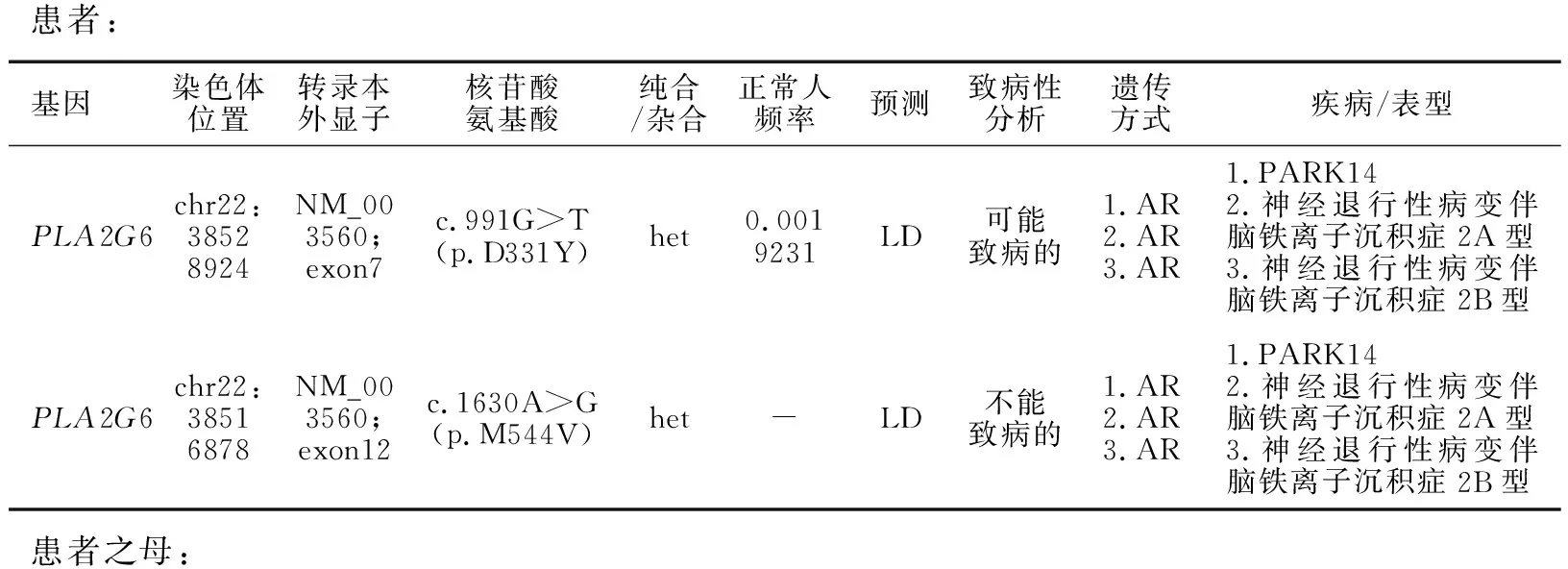
患者:基因染色体位置转录本外显子核苷酸氨基酸纯合/杂合正常人频率预测致病性分析遗传方式疾病/表型PLA2G6chr22:38528924NM_003560;exon7c.991G>T(p.D331Y)het0.0019231LD可能致病的1.AR2.AR3.AR1.PARK142.神经退行性病变伴脑铁离子沉积症2A型3.神经退行性病变伴脑铁离子沉积症2B型PLA2G6chr22:38516878NM_003560;exon12c.1630A>G(p.M544V)het-LD不能致病的1.AR2.AR3.AR1.PARK142.神经退行性病变伴脑铁离子沉积症2A型3.神经退行性病变伴脑铁离子沉积症2B型患者之母:
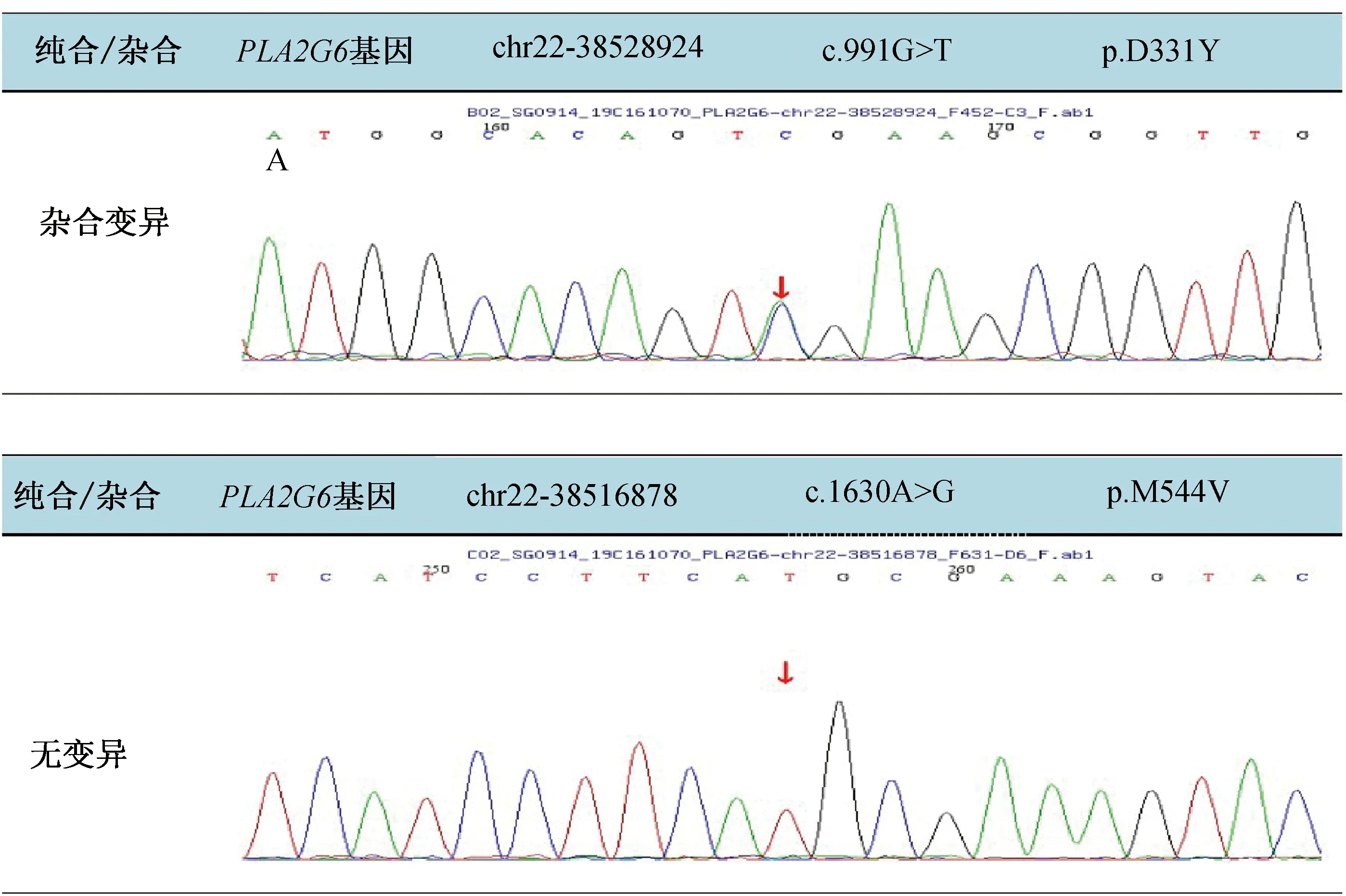
图2 患者及其母亲基因检测结果。患者携带PLA2G6基因的复合杂合突变,其第7外显子检出c.991G>T(NM_003560)杂合突变,导致第331号氨基酸由天冬氨酸变为酪氨酸(p.D331Y),第12外显子检出c.1630A>G(NM_003560)杂合突变,导致第544号氨基酸由甲硫氨酸变为缬氨酸(p.M544V),经一代测序家系验证前者来源于其母亲
2 讨 论
PLA2G6基因即磷脂酶A2第Ⅵ型基因,位于染色体22q13.1上,大小为6.0 Mb,有17个外显子。PLA2G6基因突变为常染色体隐性遗传。该基因编码非Ca2+依赖性的磷脂酶A2(iPLA2),该酶主要有两种形式:iPLA2-A和iPLA2-β。iPLA2-β是主要的功能蛋白[3]。iPLA2-β在脑内高表达,在树突和轴突末梢中含量丰富[4]。iPLA2-β可通过催化水解磷脂的第二位羟基,释放游离脂肪酸和溶血磷脂,并在磷脂膜重塑、信号转导、内质网应激介导的细胞凋亡的过程中发挥重要作用[5]。PLA2G6基因突变的致病机制尚不明确。在敲除PLA2G6基因的果蝇模型中发现iPLA2-ⅥA的功能缺损会引起线粒体功能异常,导致线粒体呼吸链功能障碍、ATP合成减少及线粒体形态异常,同时导致脂质过氧化水平及活性氧水平升高[6]。PLA2G6基因突变会导致一系列的神经变性病,称为PLA2G6相关的神经变性病(PLAN)。PLA2G6不同的突变位点会导致iPLA2-β酶活性不同程度的改变,该酶活性被认为是影响PLAN临床表型的重要因素[7]。PLA2G6基因具有高度的遗传及临床的异质性,不同的突变位点、类型及人种,PLAN的临床表现也会有所不同。根据发病年龄及临床表现的不同,主要将PLAN分为以下四个亚型:婴儿型神经轴索变性(INAD)、非典型的神经轴索变性(ANAD)、成年起病的肌张力障碍-帕金森综合征(DP)及常染色体隐性遗传的早发型帕金森综合征(AREP)。INAD及ANAD发病年龄较早,通常儿童期发病,多数患者头颅MRI会出现小脑皮质萎缩及异常的脑铁沉积征象,这一表现又被分类为伴有脑铁沉积的神经变性病Ⅱ型(NBIA Ⅱ)[8]。
目前文献中报道的PLA2G6突变与PD相关的临床表型主要有三种:DP[9]、AREP[10-12]、早发型帕金森综合征(EOP)[12-13]。PLA2G6突变相关的PD多数是中青年发病,对多巴胺能药物反应良好,但容易出现多巴诱导的运动并发症,PET通常显示DAT摄取降低[10,12,14]。(1)DP:临床表现复杂,起始症状多样。主要临床表现为帕金森症状伴有肌张力障碍,随着疾病进展可出现非运动症状,如认知减退、抑郁、精神症状等。文献总结有33%的患者MRI会出现异常的脑铁沉积,并且部分患者T2WI未检出脑铁沉积,在磁敏感相(SWI)或梯度回波序列检测出脑铁沉积征象,提示在临床工作中注意完善检查序列。部分患者出现小脑萎缩,该征象可能对本病有提示意义[14]。(2)AREP:该型患者临床症状较轻,表现为典型的帕金森症状,无肌张力障碍,病程进展相对较慢。目前报道的病例MRI上未发现有异常的脑铁沉积征象[10-12]。(3)EOP:有报道[15-16]在EOP患者中检出PLA2G6突变,临床表现多样,主要临床表现为对左旋多巴反应良好的EOP,部分伴有痴呆和额颞叶萎缩,无肌张力障碍、锥体束征等,随着疾病进展可能出现认知减退、精神症状等,部分患者头颅MRI出现脑铁沉积征象。与AREP相比,EOP多无家族史[17]。
Lu等[12]通过对25例中国YOPD患者进行基因测序,在其中的5例患者基因结果中发现PLA2G6基因突变,其推测PLA2G6基因突变可能是中国YOPD的一个风险因素。PLA2G6基因在PD发病机制中的具体作用仍不明确,推测其可能与炎性反应、tau蛋白和α-突触核蛋白的错误折叠与聚集、细胞凋亡、线粒体功能障碍等相关[3]。Lin等[18]近期研究发现,iPLA-ⅥA功能缺失可导致retromer复合体功能障碍及鞘磷脂代谢失衡,造成神经酰胺水平升高,导致神经元损伤,也可能是PLA2G6相关PD的致病机制。PLA2G6基因不同的突变位点、类型及人种间,PD的临床表型亦可不同[3](表1)。近些年来,有病例报告指出PLA2G6基因突变临床上可表现为遗传性痉挛性截瘫,该研究进一步扩大了PLAN的临床表型[19-20]。随着PLA2G6突变新位点的增加,PLAN的表型逐步扩大,提示在临床工作中应该重视对PLA2G6基因的检测。
本例患者右下肢震颤起病,右侧肢体肌张力增高、右下肢少动、肌张力障碍定位在锥体外系,四肢腱反射增强定位于锥体系;便秘症状定位于自主神经。使用小量美多芭后,症状明显改善;根据MDS-PD诊断标准该患者具有运动迟缓、肌强直和静止性震颤可诊断为帕金森综合征,具有2条支持标准(对多巴胺类药物有显著的反应、明确的非对称的静止性震颤),无绝对排除标准,1条警示标准(不明原因的反射活跃),诊断很可能的PD,发病年龄<50岁,考虑YOPD可能性大。YOPD病因通常是基因、表观遗传和环境的相互作用[21]。该患者遗传因素需考虑,行基因检测发现PLA2G6的c.991G>T及c.1630A>G复合杂合突变。c.991G>T(p.D331Y)在Pubmed中已有记录,c.991G>T突变可引起线粒体功能障碍、内质网应激升高和转录异常诱导黑质致密区多巴胺能神经元早发变性,具有致病性[22]。并且,c.991G>T在中国人群中是PLA2G6的一个热点突变[23]。位点c.1630A>G(p.M544V)突变在ClinVar数据库未被查询到,本文是首次报道该位点突变,REVEL软件对其的预测结果为潜在有害,PolyPhen_2、MutationTaster和GERP+对该位点突变的预测结果为有害。通过在ClinVar上查询,该突变位点编码的氨基酸的位置位于iPLA2的Patatin功能域处,该点的突变可能会对基因产物的功能产生影响。该位点虽未经报道,但多种预测软件预测其可能对基因表达产物及功能产生有害作用。根据ACMG指南,该变异位点初步判定为临床意义未明。c.991G>T(p.D331Y)及c.1630A>G(p.M544V)均为杂合突变,前一位点突变为已报道的导致PARK14相关PD的致病基因,后者虽未经报道,但多种计算机计算法预测其可能对基因表达产物结构及功能产生有害作用,结合患者表现为PD症状、肌张力障碍,左旋多巴治疗有效,较为符合PARK14相关PD的表现。这一复合杂合突变可能是这一PLAN表型的致病基因。需要指出的是,既往报道[8,10-11]中纯合突变c.991G>T的临床表型为AREP型。本例患者的临床表型考虑为DP型,推测新突变位点c.1630A>G参与影响临床表型的可能性大。
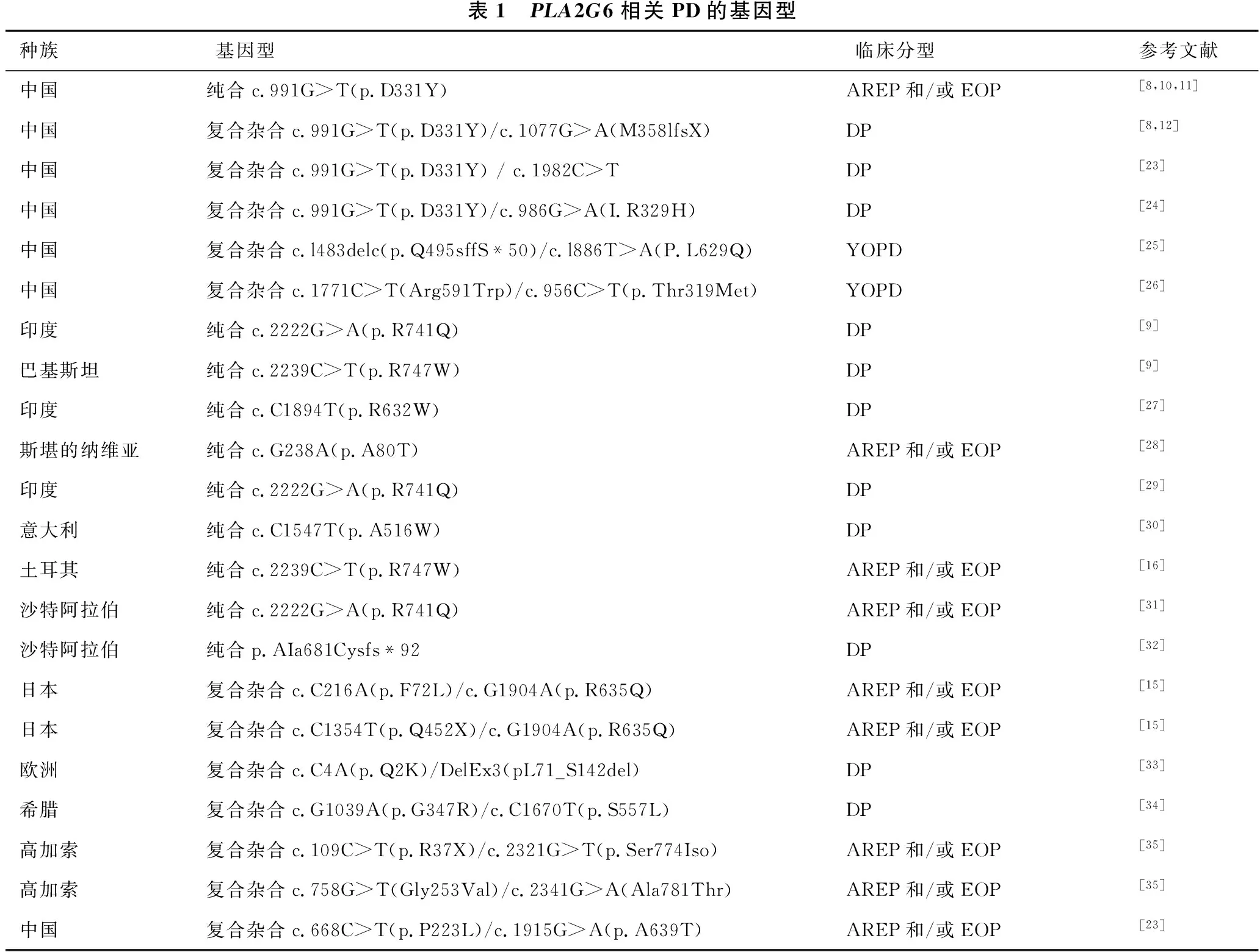
表1 PLA2G6相关PD的基因型种族基因型临床分型参考文献中国纯合c.991G>T(p.D331Y)AREP和/或EOP[8,10,11]中国复合杂合c.991G>T(p.D331Y)/c.1077G>A(M358lfsX)DP[8,12]中国复合杂合c.991G>T(p.D331Y) / c.1982C>TDP[23]中国复合杂合c.991G>T(p.D331Y)/c.986G>A(I.R329H)DP[24]中国复合杂合c.l483delc(p.Q495sffS*50)/c.l886T>A(P.L629Q)YOPD[25]中国复合杂合c.1771C>T(Arg591Trp)/c.956C>T(p.Thr319Met)YOPD[26]印度纯合c.2222G>A(p.R741Q)DP[9]巴基斯坦纯合c.2239C>T(p.R747W)DP[9]印度纯合c.C1894T(p.R632W)DP[27]斯堪的纳维亚纯合c.G238A(p.A80T)AREP和/或EOP[28]印度纯合c.2222G>A(p.R741Q)DP[29]意大利纯合c.C1547T(p.A516W)DP[30]土耳其纯合c.2239C>T(p.R747W)AREP和/或EOP[16]沙特阿拉伯纯合c.2222G>A(p.R741Q)AREP和/或EOP[31]沙特阿拉伯纯合p.AIa681Cysfs*92DP[32]日本复合杂合c.C216A(p.F72L)/c.G1904A(p.R635Q)AREP和/或EOP[15]日本复合杂合c.C1354T(p.Q452X)/c.G1904A(p.R635Q)AREP和/或EOP[15]欧洲复合杂合c.C4A(p.Q2K)/DelEx3(pL71_S142del)DP[33]希腊复合杂合c.G1039A(p.G347R)/c.C1670T(p.S557L)DP[34]高加索复合杂合c.109C>T(p.R37X)/c.2321G>T(p.Ser774Iso)AREP和/或EOP[35]高加索复合杂合c.758G>T(Gly253Val)/c.2341G>A(Ala781Thr)AREP和/或EOP[35]中国复合杂合c.668C>T(p.P223L)/c.1915G>A(p.A639T)AREP和/或EOP[23]
本病应与多巴反应性肌张力障碍相鉴别,多巴反应性肌张力障碍主要表现儿童期起病的肢体肌张力障碍,主要表现在下肢,呈昼夜波动性,患者对左旋多巴治疗有良好和持续的反应,通常由常染色体显性遗传的GCH-1编码的三磷酸鸟苷环水解酶-1(GTP-CH-1)缺乏引起[36]。该患者临床表现与基因检测结果与之不符,暂排除此病可能。
综上所述,本例患者主要表现为PD症状、肌张力障碍,美多芭治疗有效,符合DP的特点,MRI未发现明显异常,但其未完成SWI序列,后期需完善。治疗期间暂未出现运动并发症,后期注意随访,记录患者对多巴的反应性,警惕运动并发症的出现。本例患者属于不典型的YOPD,对于不典型的YOPD患者,需考虑遗传性因素,必要时应进一步完善相关基因检查。
