微波等离子体原子发射光谱法测定偏远矿区海泡石中的主微量元素
2021-11-01周学忠谢华林
周学忠, 谢华林
(1.湖南工学院材料科学与工程学院, 湖南 衡阳 421002;2.长江师范学院材料科学与工程学院, 重庆 涪陵 408100)
海泡石是具有层状结构的含水富镁硅酸盐黏土矿物[1],由两个Si—O四面体包裹一个Mg—O八面体构成的特殊结构赋予了海泡石具有比表面积大、收缩率低、可塑性好等特征[2-4],因其极强的吸附和分散性被广泛用作脱色、净化和催化剂载体[5-7]。受成矿物质来源、成矿流体性质和矿床成因的影响,海泡石中无机元素的组成和含量存在差异,从而影响海泡石的性能和质量,特别是海泡石中所含微量毒理性元素通过食物链被人体摄入,造成毒理性元素的积蓄危害。因此,建立快速、准确地测定海泡石中主量和微量元素的分析方法,对于海泡石的开发利用及其质量等级评估均具有十分重要的意义。
目前,有关海泡石的研究主要集中于结构和性能的研究[8-13],对于海泡石中无机元素的检测多参照国家标准《硅酸盐岩石化学分析方法》(GB/T 14506—2010)进行测定,制约了海泡石的发展应用。GB/T 14506—2010中采用分光光度法测定Ti和Fe,容量滴定法测定Ca、Al、Mg,原子吸收光谱法(AAS)测定Na和K。其中,分光光度法和容量滴定法操作复杂繁琐且灵敏度低;AAS法虽然具有较高的灵敏度和较低的检出限,但该方法属于单元素测定方法,对于多元素的分析不具有快速检测能力,而且对于一些难电离元素的分析其原子化效率低,采用易燃或氧化性气体存在安全隐患。例如,宋帅娣等[14]利用王水提取海泡石中的微量As,采用氢化物发生-原子荧光光谱法(AFS)进行测定,但原子荧光淬灭效应以及散射光的干扰限制了AFS的推广应用,只能针对某些特定元素的测定,不具有通用性;王力强等[15]利用敞口酸溶对海泡石进行微波消解,采用电感耦合等离子体发射光谱法(ICP-OES)测定7种主量成分;张楠等[16]利用二元混合酸对海泡石进行微波消解,采用电感耦合等离子体质谱法(ICP-MS)测定15种稀土元素。然而,以电感耦合等离子体(ICP)为激发光源的ICP-OES和ICP-MS虽然具有线性范围宽、检出限低、灵敏度高且进行多元素连续测定的高通量特征,但ICP在高温激发过程中会产生成大量谱线干扰,持续使用以高纯氩气为工作气的ICP需要配备专用供气设备,对于偏远矿区和生产现场将面对气体采购运输困难和使用不便等难题。
微波等离子体原子发射光谱法(MP-AES)采用氮气为等离子体工作气,通过使用磁场耦合微波能量从而激发等离子体,实现了高效激发与能量的耦合,形成稳定的低温等离子体,具有运行稳定、光谱干扰少、适用性强等优势得到了广泛应用[17-23]。本文以硝酸-盐酸-氢氟酸三元混合酸为消解试剂对海泡石进行微波消解,采用MP-AES测定其主量元素Mg、Al、Ca、Fe、K、Na和微量元素Cu、Zn、Mn、Pb,通过选择分析波长并结合快速线性干扰校正(FLIC)技术消除光谱干扰,以期为海泡石中多种元素的快速准确检测提供一种新方法。
1 实验部分
1.1 仪器及工作条件
Agilent 4200微波等离子体原子发射光谱仪(MP-AES),配备惰性氧化铝进样炬管、OneNeb惰性雾化器、惰性双通道雾化室、4107氮气发生器和SPS-3自动进样器(美国Agilent公司);MARS X微波消解系统(美国CEM公司);Milli-Q超纯水机(美国Millipore公司)。
MP-AES优化后的操作条件为:雾化气流速0.3~0.95L/min;泵速10r/min;读数时间3s;重复次数3次;稳定时间10s;取样吸入延迟时间45s;冲洗时间120s。各元素分析波长为:Mg 285.213nm,Al 396.152nm,Ca 393.366nm,Fe 371.993nm,K 769.892nm,Na 589.592nm,Cu 327.395nm,Zn 213.857nm,Mn 403.076nm,Pb 405.781nm。
1.2 标准溶液和主要试剂
1000mg/L的Mg、Al、Ca、Fe、K、Na、Cu、Zn、Mn、Pb单元素标准溶液:购自国家有色金属及电子材料分析测试中心;1000mg/L的Lu单元素内标溶液:购自国药集团化学试剂有限公司。
65%的硝酸、37%的盐酸、40%的氢氟酸:购自德国Merck公司。
实验用水为Milli-Q超纯水。
1.3 实验样品
海泡石成分分析标准物质(GBW07138):购自中国地质调查局天津地质调查中心。
海泡石实际矿物样品来自湖南浏阳。
1.4 样品处理
海泡石经细碎制成粒径≤74μm的样品,在105℃温度下烘干2h后保存于干燥器内备用。准确称取已烘干的海泡石样品0.2g于微波消解罐中,依次加入2mL超纯水、1mL硝酸、3mL盐酸、1mL氢氟酸,浸泡过夜后进行微波消解。设置微波消解最大功率1600W,梯度升温程序为:5min升温至120℃,3min升温至150℃并保持5min,5min升温至185℃并保持15min。消解完成后冷却至室温,用超纯水将澄清透明消解溶液转移至100mL聚乙烯容量瓶中定容,待测。同时制备空白溶液。
1.5 仪器测定
分别配制不同浓度梯度的系列混合标准溶液,在优化的MP-AES操作条件下进行测定,利用分析元素与内标元素谱线强度的比值(相对谱线强度)所对应的分析元素标准溶液浓度建立校准曲线,同时对空白溶液和样品溶液进行测定,所有测试溶液均在线加入1mg/L的内标溶液。根据样品溶液中分析元素的相对谱线强度,利用校准曲线计算样品溶液中分析元素的含量。所有数据均采用MP-AES自带的MP Expert 软件进行自动处理。
1.5 校准曲线
采用MP-AES测定不同浓度的系列混合标准溶液,以分析元素与内标元素的相对信号强度所对应的浓度建立校准曲线,利用MP Expert自动得到分析元素的线性数据。
2 结果与讨论
2.1 样品处理方法的选择
实验选择采用海泡石成分分析标准物质(GBW07138)确定样品处理方法。采用碱熔法完全可以分解海泡石,但由于加入碱性熔剂无法对样品中的碱金属元素进行测定,且高温熔融会导致元素损失,因此本实验采用酸来消解海泡石样品。硝酸作为最常用的消解试剂,单独使用时其氧化能力不足以破坏海泡石的Si—O四面体结构[24]。通过图1可以看出,所有分析元素的提取率均偏低;采用硝酸-氢氟酸二元混合酸为消解试剂经微波消解虽然能完全分解海泡石,但样品溶液中低浓度Pb表现出高背景和偏低提取率;采用硝酸-盐酸-氢氟酸三元混合酸为消解试剂,盐酸所提供的氯离子能与多数金属离子配位,表现为样品更易溶解,样品溶液的稳定性更好,所有分析元素的提取率高(≥95%)。因此,本实验选择硝酸-盐酸-氢氟酸为消解试剂,经微波消解后无需赶酸,具有操作简单、分析快速、溶液稳定性好等优势。

图1 不同消解体系对分析元素的提取率Fig.1 Extraction rate of elements in different digestion systems
2.2 分析波长的选择
MP的等离子体温度比氩气低2000~3000K,受激发温度的限制,对于高电离能的元素难以实现完全电离,同时,MP不能有效诱导样品的热分解[17]。因此,MP所形成的发射光谱线比ICP少,并且多数光谱线以原子线的形态存在,虽然减少了大量光谱重叠干扰和易电离元素的电离干扰,但可用于分析的光谱线也变少。本实验利用MP Expert自动调用分析元素的预设波长,根据元素谱线库凸显的潜在干扰,选择灵敏度高、光谱重叠干扰少的光谱线作为分析波长。
Mg的灵敏谱线分别为285.213nm、279.553nm和383.829nm,其中285.213nm和383.829nm是Mg原子线,279.553nm是Mg离子线,通过扫描发现,在Mg的279.553nm处,Mn的两条高响应强度原子线279.482nm和279.827nm对Mg产生严重干扰。而对于Mg的两条原子线285.213nm和383.829nm均无潜在干扰,但相同浓度的Mg在285.213nm的灵敏度比383.829nm高约40倍。因此,实验选择Mg的分析波长为285.213nm。Al在396.152nm和394.401nm处有两条高灵敏原子线,并且都没受到光谱干扰,二者均能用于测定Al,实验选择396.152nm为Al的分析波长。Ca在393.366nm处有灵敏的离子线,在422.673nm处有灵敏的原子线,对于Ca在422.673nm处的原子线与Fe的三条原子线422.545nm、422.417nm、422.743nm相重叠,而Fe在海泡石中属于主量元素,故实验选择Ca的分析波长为393.366nm。Fe的高灵敏离子线在259.940nm,高灵敏原子线在371.993nm,二者受到来自稀土元素的干扰几乎可以忽略不计,在ICP-OES分析中,通常选择259.940nm为分析波长,但在MP-AES分析中,受低温等离子体限制,Fe的原子线响应强度比离子线高约一个数量级,故实验选择Fe的原子线371.993nm为分析波长。K的灵敏谱线在766.491nm和769.892nm处,相同浓度的K在766.491nm响应强度是769.892nm的2倍左右,而Na的灵敏谱线在588.595nm和589.592nm处,相同浓度的Na在588.595nm的响应强度是589.592nm处的2倍左右,但由于K在766.491nm会受到Lu原子线766.434nm的干扰,Na在588.595nm会受到Mo原子线588.831nm的干扰,实验选择K和Na的分析波长分别为769.892nm和589.592nm。
Cu的高灵敏谱线为两种原子线324.754nm和327.395nm,相同浓度的Cu在324.754nm的响应是327.395nm的2倍,但324.754nm受到来自Fe谱线324.596nm、324.696nm和324.820nm的干扰以及La谱线324.935nm的干扰,而Cu在327.395nm的灵敏线也会受到来自Ca原子线327.467nm的潜在干扰,实验选择干扰相对较少的327.395nm为Cu的分析波长。Zn有两条高灵敏原子线213.857nm和472.215nm。与Cu的光谱干扰相似,Zn的两条原子线均存在潜在干扰,Fe的原子线和V离子线对Zn原子线213.857nm产生干扰,而La的离子线对Zn原子线472.215nm产生干扰,由于Zn在213.857nm的灵敏度比472.215nm高60倍,实验选择Zn的分析波长为213.857nm。Mn在403.076nm处有灵敏的原子线,在259.372nm处有较灵敏的离子线,Fe原子线对Mn原子线403.076nm和离子线259.372nm均存在轻微干扰,同时La原子线403.169nm对Mn原子线403.076nm构成较强干扰,但由于样品溶液中La的浓度很低,对Mn的干扰可以忽略不计,实验选择403.076nm为Mn的分析波长。Pb有多种原子线可供选择,但几乎所有可用于测定的原子线均存在谱线重叠干扰,因此,实验选择最灵敏原子线405.781nm为Pb的分析波长。
2.3 干扰及校正
通过选择分析元素的谱线波长消除了大量谱线重叠干扰,但仍然存在少量此类干扰;受基质组成以及MP光源发射杂散光的影响,所有分析元素都存在背景干扰,与谱线重叠干扰共同作用构成了光谱干扰[25]。本实验选择FLIC技术通过分别测定空白溶液、分析元素标准溶液和干扰元素标准溶液的响应值来进行数据建模,使用高级光谱建模技术对所存在的光谱干扰进行校正,采用数学方式将背景干扰以及谱线重叠干扰从原始光谱信号中分离,得到分析元素的FLIC模型,从而实现了准确测定分析元素的响应信号[26-27]。图2为Cu在327.395nm的FLIC校正模型,分别选择空白溶液和100mg/L的Ca标准溶液作为干扰物,1mg/L的Cu标准溶液作为分析物进行数据建模,Cu的FLIC模型可完全校正背景干扰以及来自Ca原子线327.467nm的谱线重叠干扰。

图2 快速线性干扰校正(FLIC)模型校正Cu(327.395nm)的光谱干扰Fig.2 Correction of spectral interference of Cu (327.395nm) by fast linear interference correction (FLIC) model
针对不同溶液基质差异所产生的基体效应,采
用内标法补偿谱线强度变化[28-30]。通过在所有测试溶液中加入1mg/L的Lu为内标元素进行校正。选择Lu的分析波长为261.542nm,利用分析元素与内标元素谱线的绝对强度比值来进行定量分析。实验结果表明,加入内标元素后,稳定了分析元素的相对信号强度,所有元素在2h内连续测定12次(每10min测定一次)的相对标准偏差(RSD)均小于3%,校正了基体效应。
2.4 分析方法评价
2.4.1方法检出限
取空白溶液连续测定11次,计算各元素的标准偏差,以3倍标准偏差所对应的浓度为分析元素的检出限(LOD),以空白溶液测定值的10倍标准偏差所对应的浓度为分析方法的测定下限,以校准曲线线性范围内的最高浓度为测定上限。从表1可以看出,所有元素的线性关系良好(线性相关系数≥0.9996),测定范围能达到3~5个数量级,分析元素的检出限为0.19~14.6μg/L。
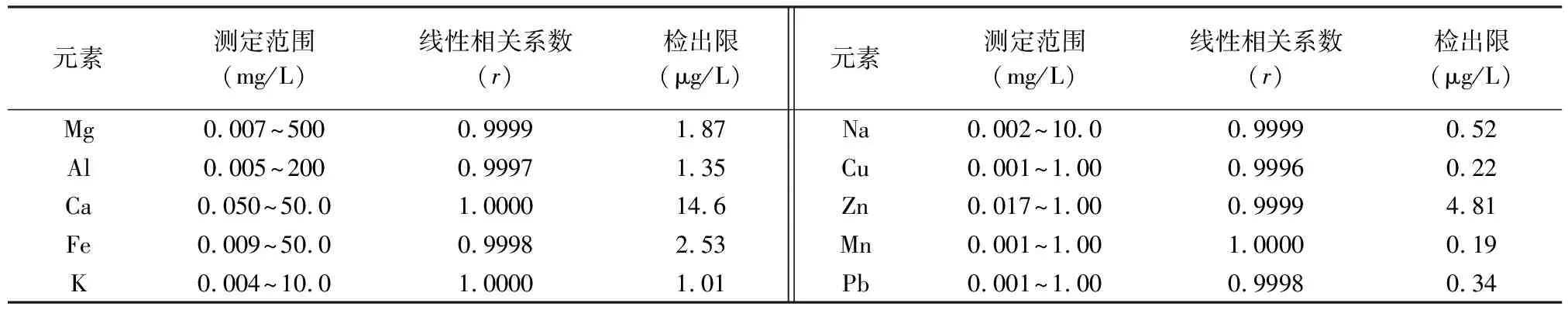
表1 分析元素的校准数据和检出限
2.4.2方法准确性和精密度
实验选择海泡石成分分析标准物质(GBW07138)评价方法的准确性和精密度,采用MP-AES重复测定6次,结果见表2。可以看出,所有分析元素的测定结果与标准值的相对误差均在-5.0%~6.7%之间,相对标准偏差(RSD)在1.4%~5.5%之间,均没有超出中国地质矿产行业标准DZ/T 0130.1—2006规定的允许限范围,验证了方法的准确性好,精密度高。

表2 MP-AES测定海泡石标准参考物质(GBW07138)的分析结果(n=6)
2.4.3海泡石样品分析
选取采自湖南浏阳的两个海泡石样品(样品编号A、B),采用本文方法对每个样品重复测定6次,采用国家标准方法GB/T 14506—2010进行对比分析,结果见表3。两种方法对比分析结果的相对误差在-4.2%~7.3%之间,RSD为2.2%~8.0%,参照中国地质矿产行业标准DZ/T 0130.1—2006,所有元素的相对误差和RSD均符合要求,表明本方法能满足实际样品中主微量元素的分析测试需求。

表3 采用MP-AES测定海泡石样品的分析结果(n=6)
3 结论
利用MP-AES对海泡石中的主量元素和微量元素进行测定,以硝酸-盐酸-氢氟酸为混合酸经微波消解能彻底分解海泡石样品,加快了样品的处理速度,提高了样品溶液的稳定性。采用FLIC建模校正光谱干扰,利用内标元素Lu校正基体效应,除Ca元素的检出限(14.6μg/L)稍高外,其余分析元素的检出限均小于5.0μg/L。
本方法具有分析运行稳定、光谱干扰少、线性动态范围宽、适用性强的优势,解决了偏远地区地质研究现场和移动实验室气体采购或运输不便的难题,并能推广应用于其他硅酸盐基质的岩石样品分析。
