通宣理肺丸HPLC指纹图谱建立及7种成分测定
2021-10-26张国强邱国玉石晓峰
李 运, 王 苗, 张国强, 邱国玉, 石晓峰, 戴 忠
(1.兰州市食品药品检验检测研究院/甘肃省种植中药材外源性污染物监测工程研究中心,甘肃 兰州 730000;2.兰州大学药学院,甘肃 兰州 730000;3.甘肃中医药大学药学院,甘肃 兰州 730000;4.甘肃省医学科学研究院,甘肃 兰州 730050;5.中国食品药品检定研究院,北京 100050)
通宣理肺丸由紫苏叶、前胡、桔梗等11味药材组成[1],方中君药紫苏叶、麻黄辛温宣肺,发散风寒;臣药前胡、桔梗辅助君药宣通肺气,杏仁润肺止咳,半夏、陈皮、茯苓燥湿化痰;佐以黄芩清肺以防肺气郁久化热,枳壳行气消痰;甘草为使药,调和诸药,全方共奏通宣理肺之功效,临床应用较广,但现行质量标准不能较全面的对其质量进行控制。因此,需借鉴指纹图谱[2]、特征图谱[3]、多成分测定[4-5]、数理统计[6]等方法,充分体现中药整体或部分化学特性的谱图,最大限度地表征药效相关成分或特征成分[7],从而在中药质量控制与评价中发挥重要的作用。
近年来,有学者对通宣理肺丸中橙皮苷、柚皮苷[8]、黄芩苷[9]、盐酸麻黄碱[10]、白花前胡甲素、白花前胡乙素[11]含量进行了测定;姚蓉等[12]筛查了前胡投料情况,并对其特征成分含量进行测定;孙国祥等[13]采用毛细管区带电泳,建立全方指纹图谱。本实验在前期报道及刘昌孝院士[14-15]提出“中药质量标志物”概念的基础上,选择与中药质量特性密切相关的标志物,建立通宣理肺丸HPLC指纹图谱,并对甘草苷、柚皮苷、橙皮苷、新橙皮苷、黄芩苷、甘草酸、白花前胡甲素含量进行测定,以期为该制剂质量控制提供参考。
1 材料
1.1 仪器 Waters e2695液相色谱仪(美国Waters公司);SBL-10DT超声波恒温清洗机(宁波新芝生物科技股份有限公司);XSE205DU电子天平(瑞士梅特勒-托利多公司);Milli-Q IQ7000超纯水机(美国默克密理博公司);ChemPattern化学计量学软件[科迈恩(北京)科技有限公司]。
1.2 试剂与药物 甘草苷(批号111610-201908,纯度95%)、柚皮苷(批号110722-201815,纯度91.7%)、橙皮苷(批号110721-202019,纯度95.3%)、新橙皮苷(批号111857-201804,纯度99.4%)、甘草酸铵(批号110731-201619,纯度96.2%)对照品均购自中国食品药品检定研究院;黄芩苷(批号8369,纯度96.2%)、白花前胡甲素(批号5636,纯度99.2%)对照品均购自上海诗丹德生物技术有限公司。通宣理肺丸(大蜜丸)31批,编号S1~S31,具体见表1。乙腈、甲醇为色谱纯;其他试剂为分析纯;纯化水为实验室自制。
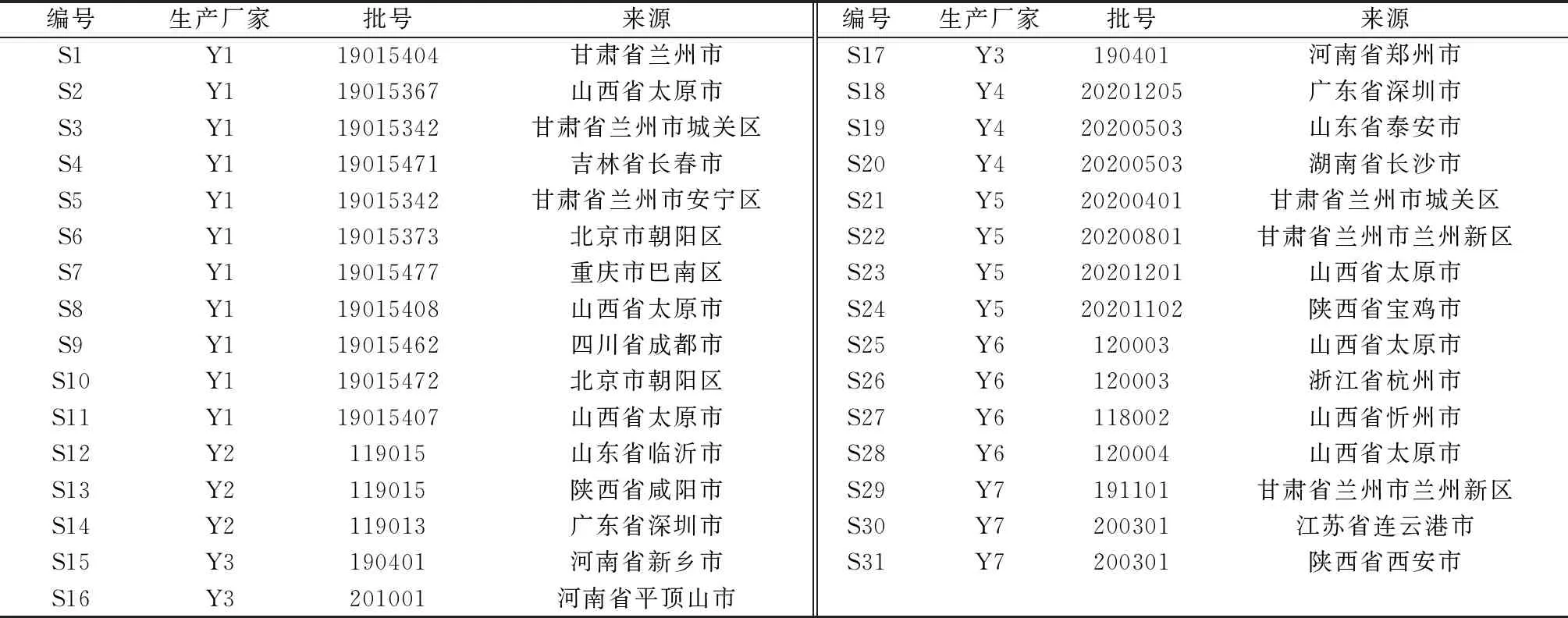
表1 样品信息
2 方法与结果
2.1 色谱条件 CAPCELL PAK C18MG色谱柱(250 mm×4.6 mm,5 μm);流动相水(含0.1%甲酸)(A)-80%乙腈(B),梯度洗脱(0~5 min,15%B;5~10 min,15%~20%B;10~20 min,20%~25%B;20~30 min,25%B;30~40 min,25%~48%B;40~45 min,48%~70%B;45~55 min,70%~90%B;56~60 min,90%~15%B);体积流量1.0 mL/min;柱温30 ℃;检测波长250 nm;进样量10 μL。
2.2 溶液制备
2.2.1 对照品溶液 精密称取各对照品适量,置于10 mL量瓶中,甲醇溶解并定容,即得(甘草苷、柚皮苷、橙皮苷、新橙皮苷、黄芩苷、甘草酸、白花前胡甲素质量浓度分别为1.003 2、0.989 4、0.582 3、1.064 6、0.575 3、1.335 3、1.174 5 mg/mL),4 ℃下保存备用。
2.2.2 供试品溶液 取样品(S3)适量,剪碎,混匀,取约1 g,精密称定,加硅藻土1 g,研匀,置于150 mL锥形瓶中,精密加入70%乙醇25.00 mL,称定质量,超声提取30 min,静置冷却,70%乙醇补足减失的质量,0.45 μm微孔滤膜过滤,取续滤液,即得。
2.2.3 阴性样品溶液 按处方比例及工艺,分别制得缺甘草,缺陈皮,缺陈皮、枳壳,缺黄芩,缺前胡,缺甘草、黄芩、前胡、陈皮、枳壳的阴性样品,按“2.2.2”项下方法制备,即得。
2.3 HPLC指纹图谱建立
2.3.1 精密度试验 取样品(S3),按“2.2.2”项下方法制备供试品溶液,在 “2.1”项下色谱条件进样测定6次,以18号峰(黄芩苷)为参照,测得各共有峰相对保留时间 RSD为0.052%~0.501%,相对峰面积RSD为0.856%~2.650%,表明仪器精密度良好。
2.3.2 重复性试验 取样品(S3),按“2.2.2”项下方法平行制备6份供试品溶液,在“2.1”项下色谱条件进样测定,以18号峰(黄芩苷)为参照,测得各共有峰相对保留时间RSD为0.026%~1.000%,相对峰面积RSD为1.017%~2.498%,表明该方法重复性良好。
2.3.3 稳定性试验 取样品(S3),按“2.2.2”项下方法制备供试品溶液,于0、2、4、8、12、24 h在“2.1”项色谱条件下进样测定,以18号峰(黄芩苷)为参照,测得各共有峰相对保留时间RSD为0.035%~1.638%,相对峰面积RSD为1.005%~2.826%,表明溶液在24 h内稳定性良好。
2.3.4 图谱生成 取31批样品(S1~S31),按“2.2.2”项下方法制备供试品溶液,在“2.1”项下色谱条件进样测定,结果见图1,将所得数据导入ChemPattern软件,设置积分条件(斜率0.01,最小相对峰高0.1%,最小百分比峰面积0.25%,积分开始3,积分结束300),以75%(高四分位数)为共有峰筛选条件,采用高斯曲线模拟生成共有模式,发现39个共有峰,见图2。
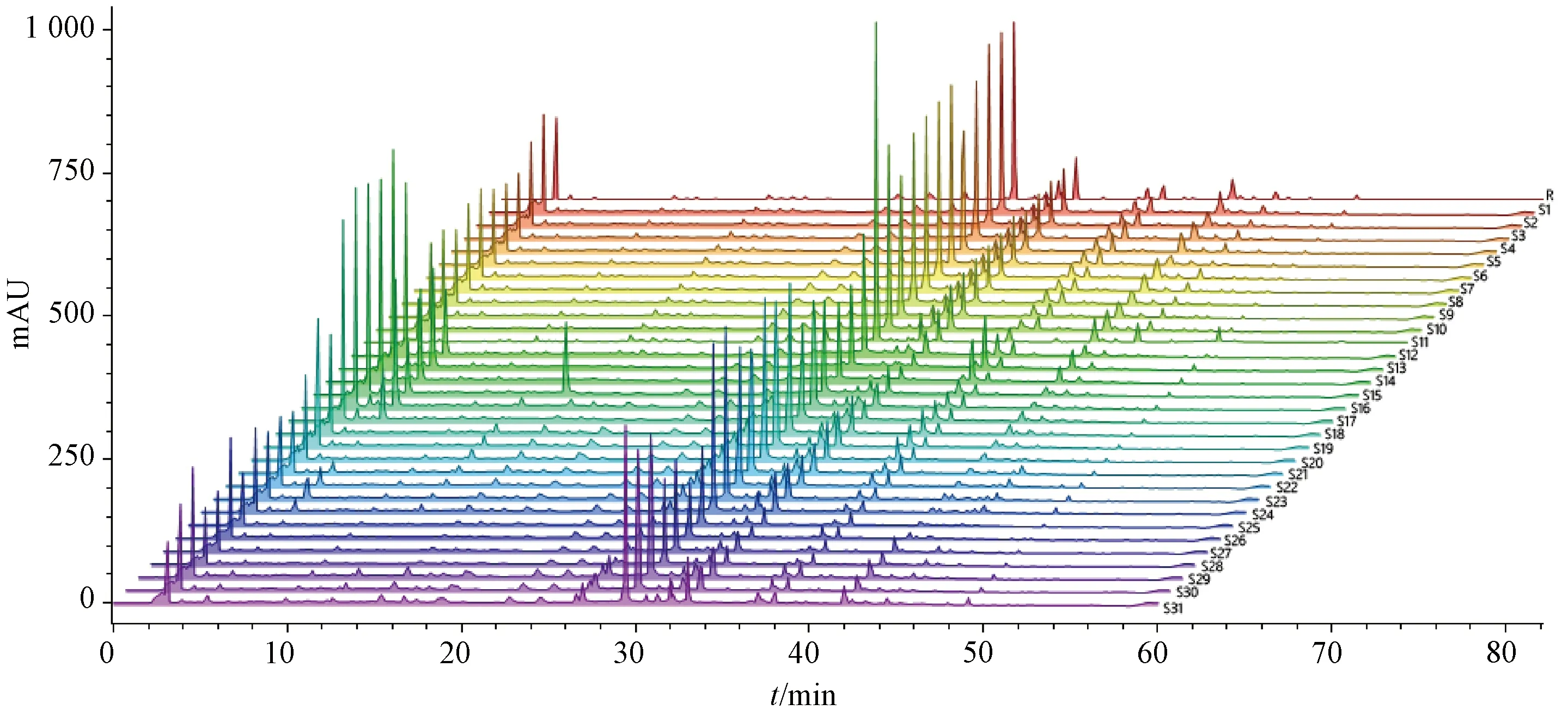
图1 31批样品HPLC指纹图谱
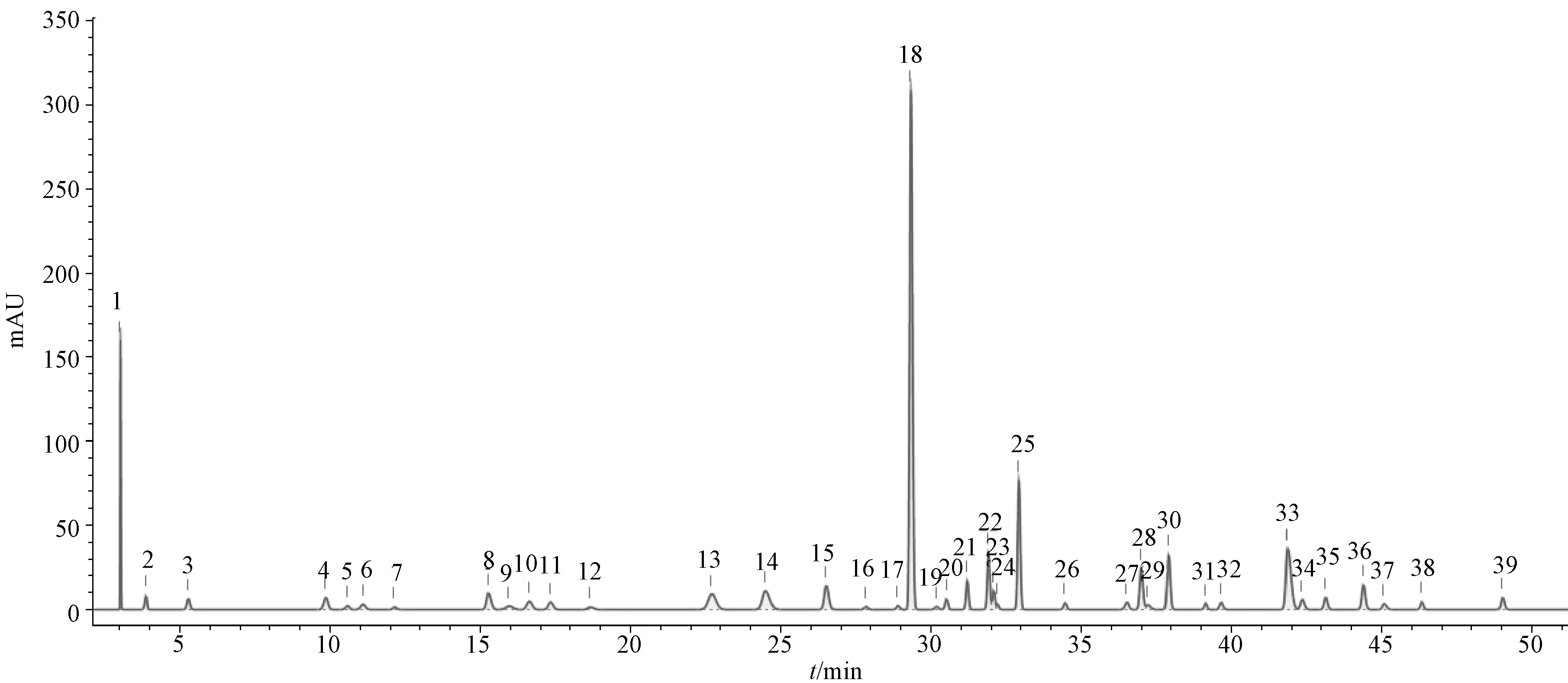
10.甘草苷 13.柚皮苷 14.橙皮苷 15.新橙皮苷 18.黄芩苷 30.甘草酸 39.白花前胡甲素 10.liquiritin 13.naringin 14.hesperidin 15. neo-hesperidin 18.baicalin 30. glycyrrhizic acid 39.praeruptorin A
2.3.5 相似度分析 采用ChemPattern软件,以HPLC指纹图谱共有模式为参照图谱,通过夹角余弦法计算各样品相似度,见表2,可知相似度为0.16~0.99,其中有77.4%大于0.95,表明不同企业样品质量差异较大,其中Y2、Y3企业有3批低于0.6,均存在部分特征峰丢失或者峰面积偏小的情况。

表2 31批样品相似度
2.3.6 聚类分析 采用ChemPattern软件将31批样品HPLC指纹图谱数据进行标准化后,以街区距离为度量,通过远邻法进行聚类分析,结果见图3。
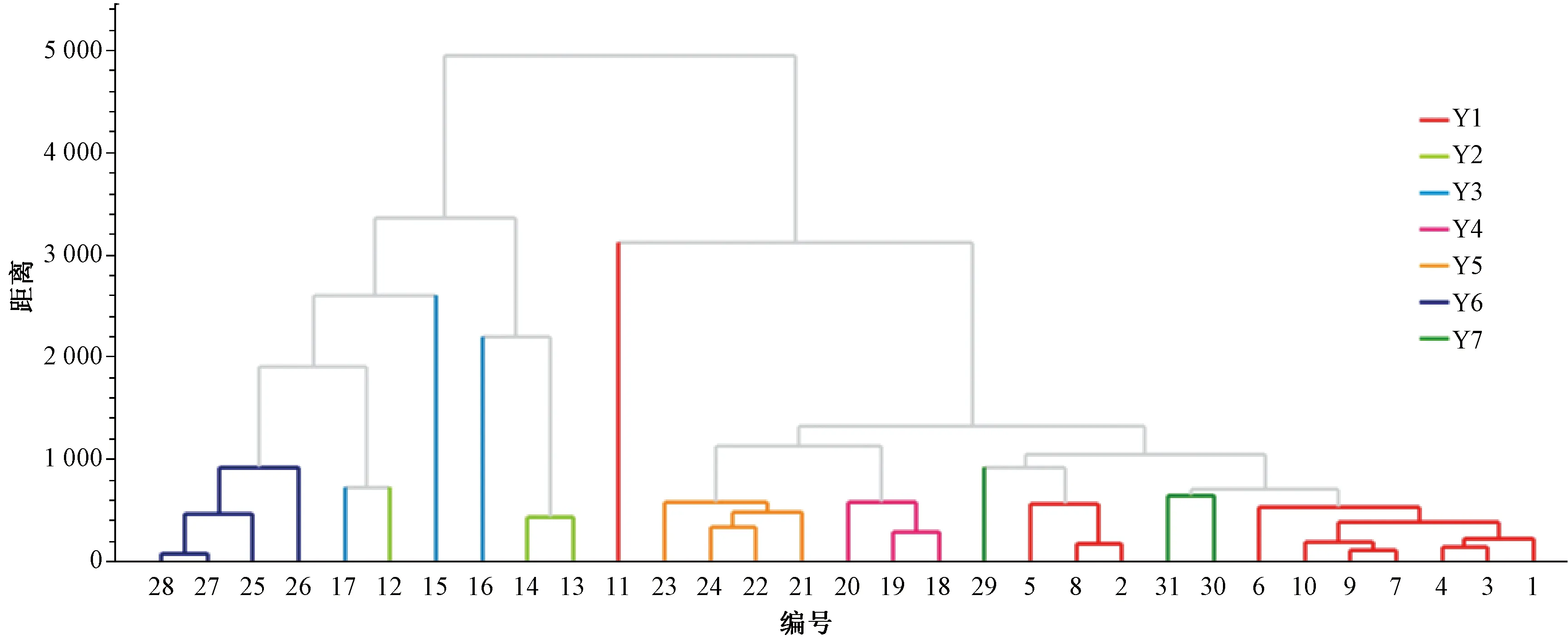
图3 31批样品聚类分析图
2.3.7 主成分分析 采用ChemPattern软件进行主成分分析,结果见图4。由此可知,第一主成分(PC1)、第二主成分(PC2)、第三主成分(PC3)贡献率分别为65.13%、25.64%、3.99%,总计94.76%,能较好地反映各批样品之间的差异;峰18、3、28、1、14、33、12、25、39是主要标志性成分,与对照品图谱比对,可知峰14为橙皮苷,峰18为黄芩苷,峰39为白花前胡甲素。
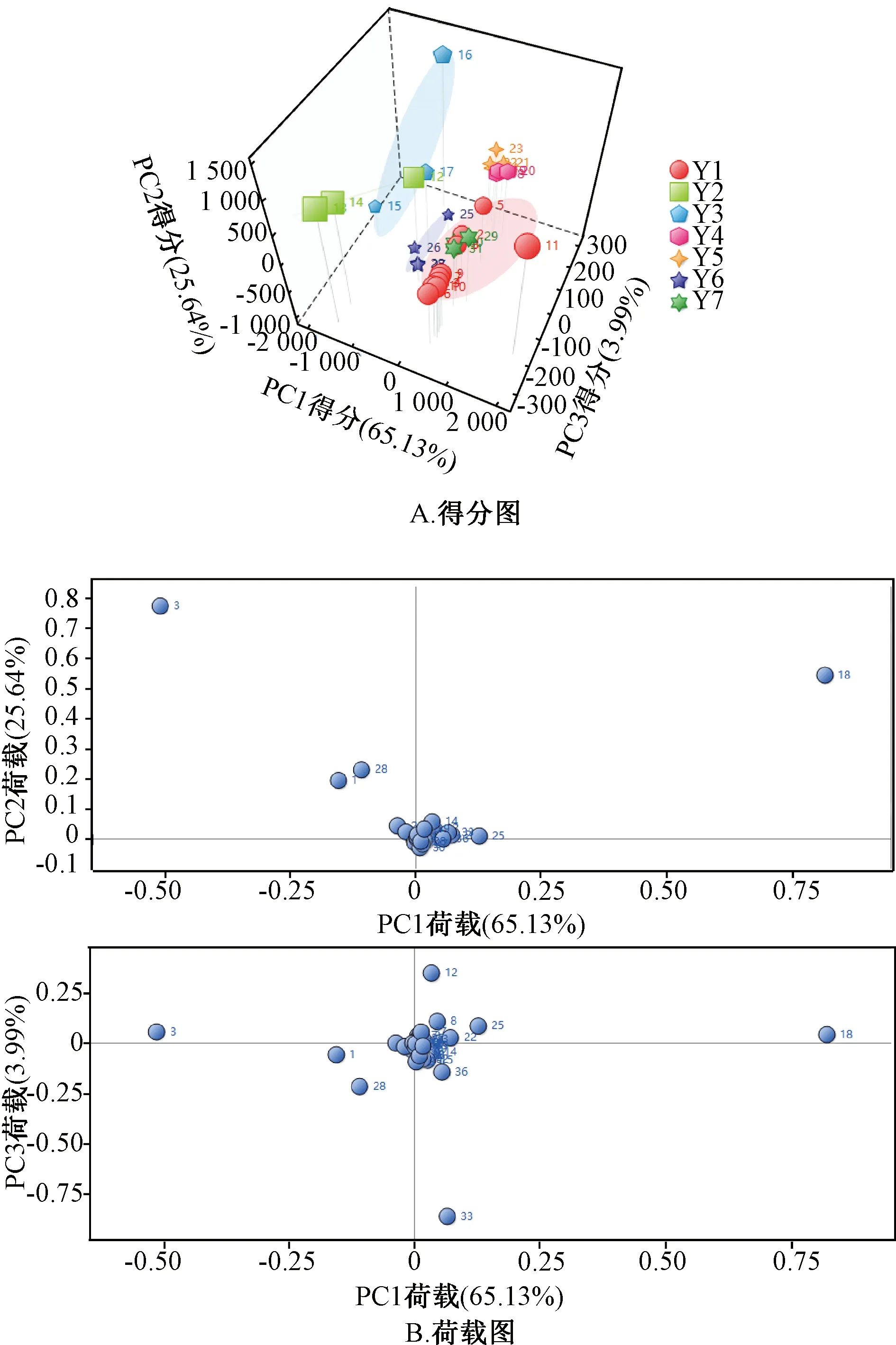
图4 31批样品主成分分析图
2.4 含量测定
2.4.1 专属性试验 取“2.2”项下对照品、供试品、阴性样品溶液,在“2.1”项下色谱条件进样测定,结果见图5。由此可知,各成分分离度良好,丸剂中其他共存物质对测定结果无干扰,理论塔板数以黄芩苷(5号峰)计,不得低于6 000。
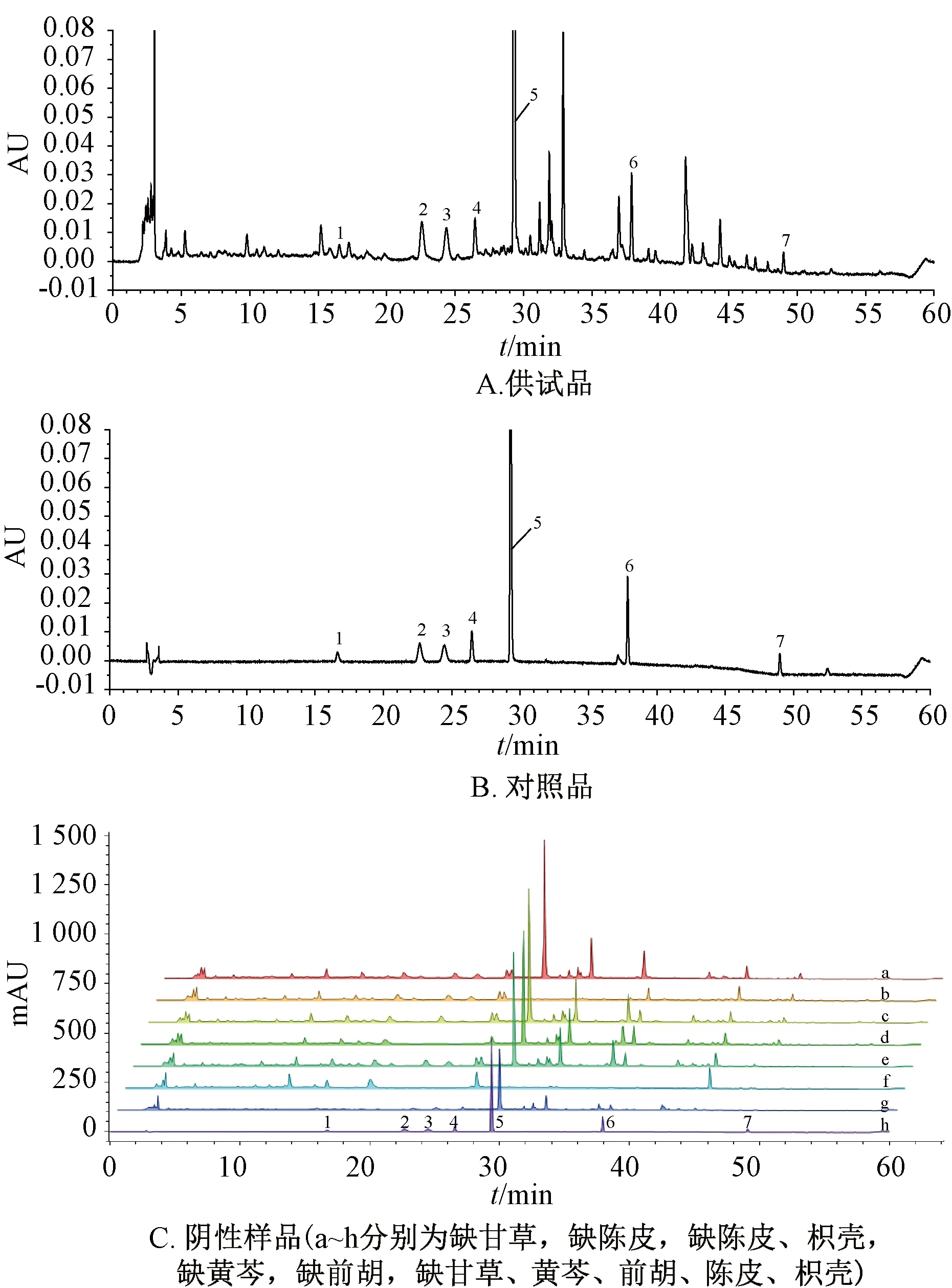
1.甘草苷 2.柚皮苷 3.橙皮苷 4.新橙皮苷 5.黄芩苷 6.甘草酸 7.白花前胡甲素
2.4.2 线性关系考察 精密吸取“2.2.1”项下对照品溶液适量,置于10 mL量瓶中,甲醇定容至刻度,摇匀,在“2.1”项色谱条件下进样测定。以对照品质量浓度为横坐标(X),峰面积为纵坐标(Y)进行回归,结果见表3,可知各成分在各自范围内线性关系良好。

表3 各成分线性关系
2.4.3 精密度试验 取“2.3.2”项下对照品溶液(甘草苷、柚皮苷、橙皮苷、新橙皮苷、黄芩苷、甘草酸、白花前胡甲素质量浓度分别为100.32、98.94、58.23、106.46、57.53、133.53、117.45 μg/mL),按“2.2.2”项下方法制备供试品溶液,在“2.1”项色谱条件下进样测定6次,测得甘草苷、柚皮苷、橙皮苷、新橙皮苷、黄芩苷、甘草酸、白花前胡甲素峰面积RSD分别为2.8%、2.3%、1.7%、0.6%、0.4%、0.8%、1.7%,表明仪器精密度良好。
2.4.4 重复性试验 取同一批样品(S3),按“2.2.2”项下方法平行制备6份供试品溶液,在“2.1”项色谱条件下别进样测定,测得甘草苷、柚皮苷、橙皮苷、新橙皮苷、黄芩苷、甘草酸、白花前胡甲素含量RSD分别为1.88%、2.15%、2.02%、1.93%、1.78%、1.45%、2.19%,表明该方法重复性良好。
2.4.5 稳定性试验 取同一批样品(S3),按“2.2.2”项下方法制备供试品溶液,于0、2、4、8、12、24 h在“2.1”项色谱条件下进样测定,测得甘草苷、柚皮苷、橙皮苷、新橙皮苷、黄芩苷、甘草酸、白花前胡甲素峰面积RSD分别为2.49%、2.32%、2.00%、2.12%、1.05%、1.24%、2.08%,表明溶液在24 h内稳定性良好。
2.4.6 加样回收率实验 精密称取各成分含量已知的样品(S3)0.5 g,共6份,加入对照品溶液,按“2.2.2”项下方法制备供试品溶液,在“2.1”项色谱条件下进样测定,计算回收率。结果,甘草苷、柚皮苷、橙皮苷、新橙皮苷、黄芩苷、甘草酸、白花前胡甲素平均加样回收率分别为99.89%、113.3%、101.2%、105.2%、93.10%、98.10%、106.6%,RSD分别为2.29%、2.58%、2.52%、1.61%、3.86%、1.25%、2.41%。
2.5 样品含量测定 取31批样品,每批2份,按“2.2.2”项下方法制备供试品溶液,在“2.1”项色谱条件下进样测定,外标法计算含量,结果见表4。
表4 各成分含量测定结果(%,n=3)
3 讨论
3.1 色谱条件优化
3.1.1 流动相 本实验考察了90%乙腈-水(含0.2%磷酸)、80%乙腈-水(含0.2%磷酸)、80%乙腈-水(含0.1%甲酸),发现80%乙腈-水(含0.1%甲酸)梯度洗脱60 min时,各成分色谱峰峰形较好。
3.1.2 检测波长 本实验采用二极管阵列检测器对各成分进行全波长扫描,综合考虑不同吸收波长处的图谱响应强度,最终确定250 nm作为检测波长,此时各成分检测灵敏度较好,干扰小。
3.2 提取条件优化 本实验考察了不同提取方法(超声、回流)、提取溶剂(70%乙醇、70%甲醇、甲醇)、提取时间(30 min、1 h、1.5 h),结合绿色分析的需要,最终确定25 mL 70%乙醇超声提取30 min作为提取条件。
3.3 含量测定分析 企业Y1、Y4、Y5、Y6、Y7样品之间的相似度均大于0.95,质量均一性良好;Y2、Y3不同批次及同一批次样品之间存在较大差异。31批样品中,甘草苷、柚皮苷、橙皮苷、新橙皮苷、黄芩苷、甘草酸、白花前胡甲素质量分数范围分别为0.001 2%~0.063 1%、0.083 8%~0.209 8%、0.007 1%~0.258 4%、0.051 4%~0.207 7%、1.43%~4.31%、0.046 4%~0.087 9%、0.013 1%~0.046 1%,差异较明显,故对通宣理肺丸质量的整体控制方法有待进一步完善。
4 结论
本实验建立了通宣理肺丸HPLC指纹图谱,并同时测定了甘草苷、柚皮苷、橙皮苷、新橙皮苷、黄芩苷、甘草酸、白花前胡甲素含量,发现指纹图谱结合数理统计能很好地从整体上表征该制剂特性,而且该方法简便准确,重复性、稳定性良好,可为其质量控制提供参考。
