光促酶催化反应设计及生物合成应用
2021-10-25张武元孙周通
张武元,袁 波,曲 戈,孙周通
(中国科学院 天津工业生物技术研究所国家合成生物技术创新中心,天津 300308)
得益于分子生物学、生物信息学、合成生物学、蛋白质工程与过程工程等学科及技术的快速发展,生物催化已成为一种在生物制药、生物能源、食品饲料与日化用品等领域中广泛使用的技术。生物催化可以改善化学反应过程的可持续性,弥补化学催化在原子经济性、区域/立体选择性、催化活性等方面的不足。当前用于生产化学品的方法过度依赖耗时、耗能和耗材的反应中间体的分离及纯化,导致大量三废排放及生产成本的增加。因此,必须通过整合符合新旧12条绿色化学准则的合成步骤来建立更清洁环保的化学品合成路线。大自然使用一种优雅而有效的策略,即利用光作为能量,并将其转换为化学能,后者随后被释放出来以促进生物体的活动。因此,光在驱动及调节生物催化反应方面的能力越来越受到重视。近些年,已报道在生物催化反应中引入光能的人工光促酶催化(photobiocatalysis)的设计与应用[1-2]。
光合作用是自然界存在最久远、反应类型最专一的复杂生物合成系统,能够利用光能将CO2和H2O转化为生命有机化合物,是生命得以延续的基础反应之一。师法自然并超越自然,基于人类社会与生命系统发展对新物质合成的迫切需求,人工设计光促生物催化反应顺应而生。已经有多种天然或人工光敏剂被引入到生物催化反应体系中,后者有望通过高效调控酶的活性来解决生物催化领域中长期存在的众多技术问题。例如,通过设计利用光活性介体[如Ru (II)-二亚胺光敏剂、硫化镉量子点等]建立光激发诱导的电子传递链体系,可直接将还原当量传递给P450酶的血红素单元[3-4]。该方法省去了P450酶依赖的复杂电子传递链及较昂贵的还原态辅酶NAD(P)H,为提升酶催化效率和经济成本问题提供了新思路。近年研究较多的过氧合酶,其结合了P450酶的催化多功能性却无须依赖辅酶及其再生循环体系,因而在催化有机化合物的氧化官能化反应领域受到重视[5]。但是该酶对H2O2极其敏感,反应体系中高浓度H2O2会引起血红素氧化分解。因此,精确控制反应体系中H2O2的浓度对过氧合酶的高效应用至关重要[6]。利用TiO2、g-C3N4等光催化剂氧化水及甲醇等小分子化合物可控地为过氧合酶原位提供H2O2,可有效解决该酶对H2O2共底物敏感性问题[7-9]。此外,通过对天然光促酶的挖掘及定向设计改造,可实现多官能团分子的制备。普林斯顿大学的Hyster发现,黄素依赖的烯还原酶在蓝绿光诱导下,跃迁到激发态的黄素可改变烯还原酶的催化功能,可对碳氢键进行选择性活化,建立起自然条件难以催化的不对称自由基环化反应[10]。
综上,人工光促酶催化在生物催化领域已经产生实质性的影响。近年也有多篇文献综述该技术在不同领域的进展[2,11-12]。本文主要选取近年典型的光促酶催化研究报道,旨在展示该技术在生物合成方面的最新应用和有待解决的技术瓶颈,并激发化学家的兴趣,为进一步开发基于光促酶催化的绿色生物催化技术提供参考。
1 光促酶催化的发展历程
1880年首次发现叶绿体是光合作用进行的场所,并对其作用机制及能量传递路径进行了一系列研究[13]。此后,光与生命交集方面的研究热情从未停止过。1949年首次发现细菌被紫外光杀死后可在可见光的诱导下恢复活性,由此揭开了DNA光解酶发现的序幕[13]。该方面的研究在2015年获得诺贝尔化学奖。20世纪末,受光催化技术的启发,生物学家通过巧妙的设计成功地将光催化氧化还原释放的电子传递给P450酶或烯还原酶[3,14],大幅度简化了冗长电子传递路径。已有报道,先后设计多项级联光催化与酶催化进行生物合成的例子[2,12]。不仅如此,近年来由于酶定向进化技术的巨大进步,可对含有FMN、FAD等辅酶的天然酶进行智能设计改造[10,15],光诱导下进行的系列反应极大地拓展了合成化学家的视野和生物合成的边界。
当前,人工设计光促酶催化的研究在生物合成中扮演着重要的角色,通过在酶促反应中以一定的方式引入光信号,使其朝着预定的目标进行。图1为近15年来有关人工光促酶催化研究的论文发表情况。光促酶催化中光以两种方式参与:(1)在体系中引入额外的光敏剂/光催化剂(本文统称光催化元件),在光激发下引发的化学反应与酶形成级联催化模式,反应过程产生的过渡态产物直接被后续的酶催化反应加以利用转化;(2)许多天然酶含有可对光响应的辅酶,如FAD、FMN、NAD(P)H等,在光的激发下,辅酶可改变原有催化模式,同时在酶三维结构的协同下,形成一系列新型的反应路径。本文将这类天然酶统称为光促酶(photoenzyme,也有文献中称其光能酶)。为方便讨论,下文分别将这两类催化方式定义为级联型光促酶催化和诱导型光促酶催化,并着重就光促酶催化在生物合成中的最新应用展开评述。
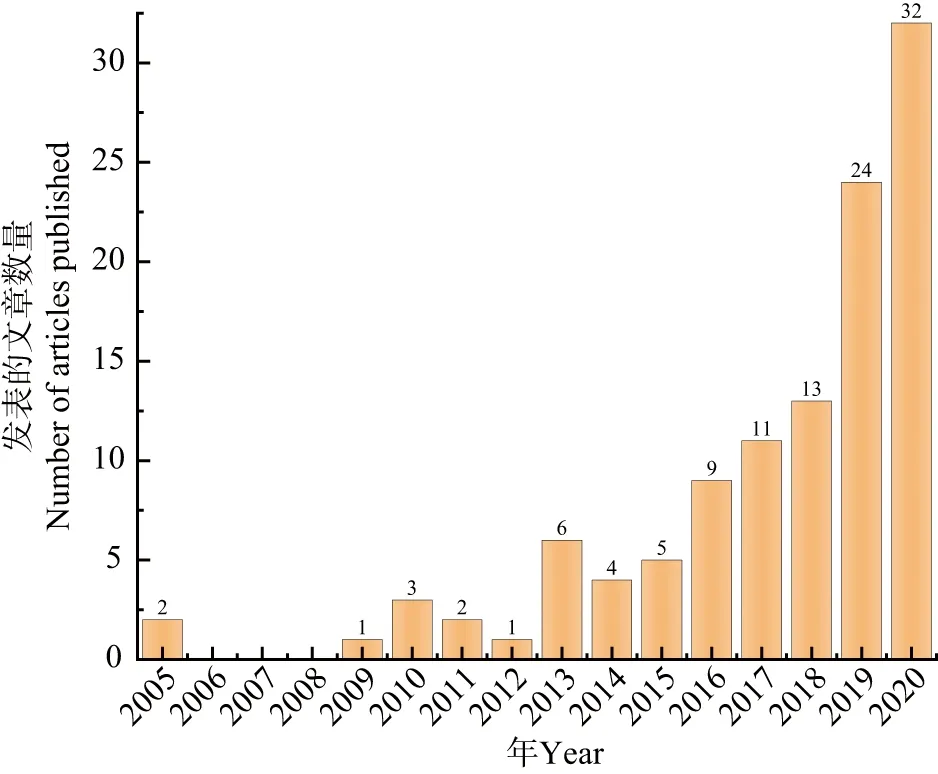
图1 2005—2020年有关光促酶催化论文发表数量(数据来自Web of Science)Figure 1 The number of published papers on photobiocatalysis from 2005 to 2020 (Data from Web of Science)
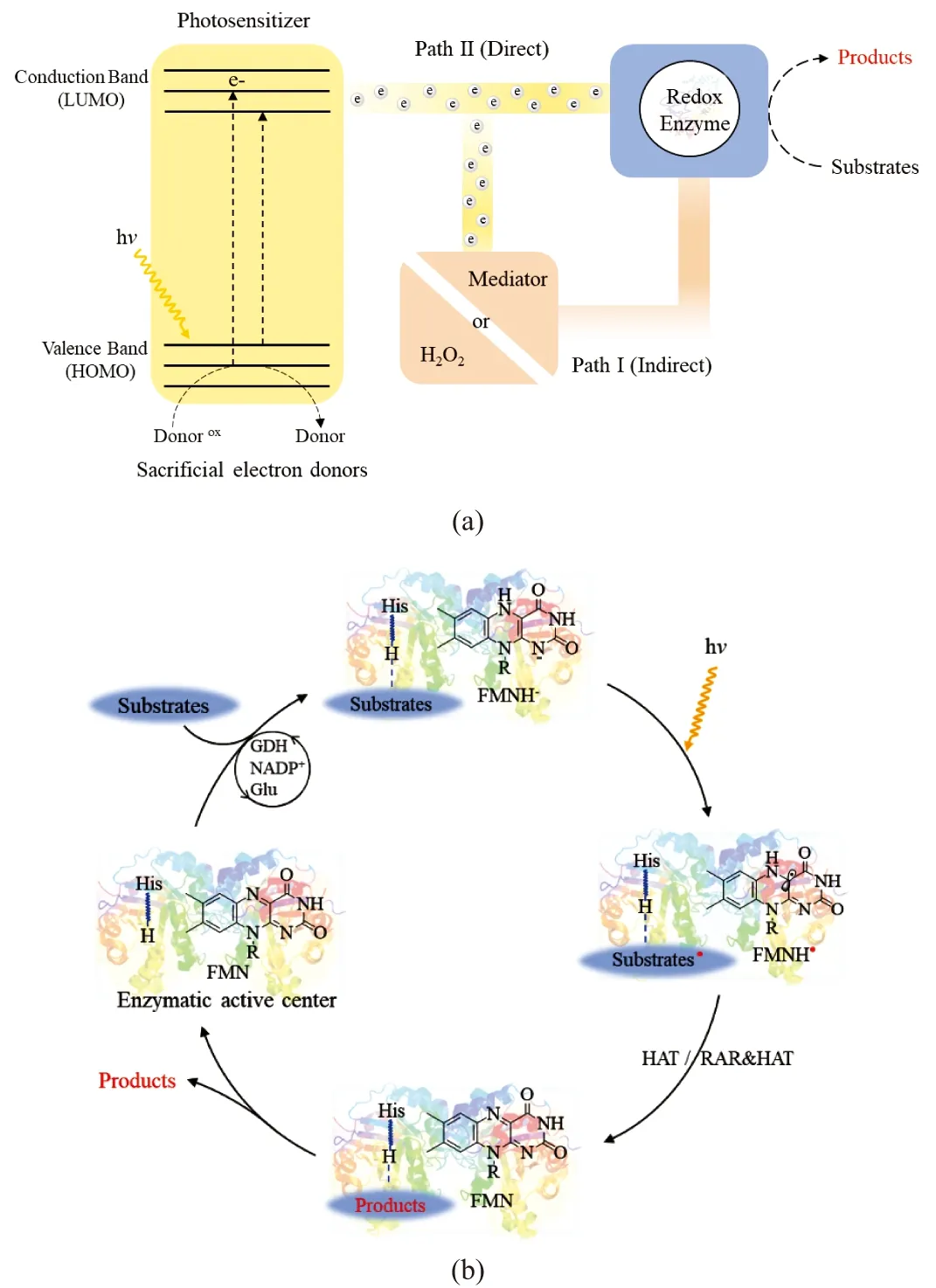
(a)级联型光促酶催化;(b)诱导型光促酶催化。 图2 光促酶催化的两种方式 Figure 2 Two types of photobiocatalytic approaches
1.1 人工光促酶催化原理
在第一类级联型光促酶催化中,人工光催化元件通过吸收可见光或者紫外光触发相邻分子的氧化还原状态。一般而言,光催化元件激发的电子可通过两种方式与酶偶联:(1)通过还原型辅因子和H2O2的光化学再生或者通过介体(Mediator)向氧化还原酶间接转移[图2(a)-Path I];(2)光激发电子直接转移到含有辅酶的氧化还原中心来激活氧化还原酶[图2(a)-Path Ⅱ]。后者可避免额外光催化再生介体的参与,因此催化过程更加简单、经济和环境友好,可最大限度地减少反应介体对底物的干扰。但是,直接光化学活化的酶往往由于光激发电子转移效率低以及副反应多导致总转化数(total turnover number,TTN)和转化频率(turnover frequency,TOF)相对较低。此外,该光促酶催化过程还面临活性氧自由基的产生和光催化剂被漂白和分解等问题[11,16]。
在第二类诱导型光促酶催化中,光直接激发含有辅酶FAD、FMN、NAD(P)H等的天然酶,无需引入额外的光催化元件。在光诱导下,辅酶直接从基态转为激发态[图2(b)]。以辅酶FMN作为活性中心的氧化还原酶催化反应为例,底物首先通过氢键作用与酶活性中心的组氨酸(histidine,His)等功能基团结合,同时氧化态FMN得到一个氢原子变成还原型FMNH-并且与底物毗邻;随后在光诱导下得到黄素自由基(FMNH·)和以底物为中心的自由基(通常以脱卤素为主)。该历程中,底物自由基直接通过氢原子转移(hydrogen atom transfer,HAT),或通过分子内/双分子间自由基加成反应(radical addition reaction,RAR)得到相应的产物和氧化态FMN;最后通过底物交换和还原型FMNH-回到静息状态,进入下一个循环[17]。以辅酶FAD和NAD(P)H为活性中心的氧化还原酶在光直接诱导型光促酶催化反应中具有相似的催化机理[10,15]。
1.2 人工光促酶催化设计中的光催化元件
有机染料[18](如卟啉、氧杂蒽衍生物、钌和铱络合物等)和一些纳米材料(如量子点、碳基纳米材料和二氧化钛等)已被广泛用作光催化元件,并由此设计了多种级联型光促酶催化方法。图3归纳了已报道的光催化元件及与之相伴的牺牲型电子供体和氧化还原介体。
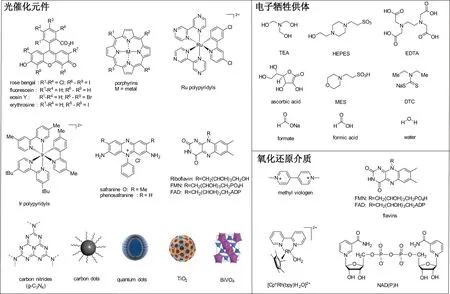
图3 光促酶体系中常用的光催化元件、牺牲电子供体和氧化还原介体Figure 3 Structures of photocatalysts,sacrificial electron donors and redox mediators commonly used for photobiocatalysis
受天然叶绿素分子中卟啉结构的特殊光化学性质启发,研究者试图设计基于卟啉结构的光催化元件[19]。例如:金属卟啉(如锌卟啉)在三重激发态下具有相对长的寿命,已被广泛用于酶催化反应体系[20-21]。杂蒽(如曙红Y和孟加拉玫瑰红等)以及藏红等属于低成本、非金属、水溶性的化合物,可吸收紫外或可见光。其结构易被功能化修饰以改进其光化学性质,产生具有单线态-三线态跃迁的光激发电子[22-23]。同理,可通过改变钌(II)和铱(III)配合物的结构,实现对其HOMO和LOMO能级调节,得到理想的光电化学性能[19]。黄素(如RF、FMN、FAD)的结构中含有芳香异咯嗪结构,具有独特的光化学和氧化还原性质,被广泛用作光促酶催化中的光催化元件。黄素在光照条件下可以作为三重态光敏剂通过系间穿越到达三重激发态发生能量转移;而由于本身的共轭结构和较宽的氧化还原电位可有效发生电子转移。这也是诱导型光促酶催化可以顺利实现的主要原因[24-25]。但是,上述有机光元件存在的普遍局限性是光漂白的发生,导致其光活性寿命较短[26]。
与有机光敏剂相比,无机纳米光催化剂在光照条件下的光漂白作用则小得多。本文主要总结几类使用频率较高的无机光催化元件。量子点(quantum dots,QD)是一种半导体纳米粒子,具有独特的光物理性质。通过精确控制QD的形状、尺寸和结构等来调控其光学性能,也可以对QD表面进行功能化改性使其更适应特定的生物酶催化环境[27-28]。二氧化钛(TiO2)禁带宽度高达3.2 eV,仅在紫外光下具有光化学响应,应用受限。通常通过对其进行掺杂改性来抑制光生电子与空穴的复合,降低TiO2禁带宽度实现可见光区域的光响应[29-30]。钒酸铋(BiVO4)具有较好的可见光响应性能,其单斜相的禁带宽度仅为2.4 eV,吸收光谱500 nm以上,化学稳定性高且安全无毒,在光促酶催化领域有广阔的应用前景[31]。碳基纳米材料如氮化碳(graphitic carbon nitride,g-C3N4)和碳量子点(carbon dot,CD)由于不含有毒重金属,生物相容性高且环境友好,以及出色及可调的光学和电化学性能在级联型光促酶催化中具有较大应用潜能[32-33]。但是,g-C3N4与氧化还原伴侣的界面相互作用较弱,在水相中的分散性较低[34]。相比之下,CD通常含丰富的羧基,水溶性好,容易通过共价键实现表面功能化[35]。
光促酶催化体系中经常需要使用电子牺牲剂再生光催化元件或使光诱导产生的空穴淬灭。常用的牺牲剂有叔胺、乙二胺四乙酸(EDTA)、有机酸(如甲酸、抗坏血酸)、含硫化合物和两性离子缓冲液等[11,36]。但多数牺牲剂的原子经济性较低,且伴随一定的副反应。因此,从原子经济性和环境方面考量,光促酶催化的理想目标是建立以甲醇作为牺牲剂的彻底氧化途径,或利用水(即反应溶剂)直接作为电子源[37]。
当光激发电子不能直接从光催化元件传递到酶活性中心时,介体的引入至关重要。后者在光催化元件和氧化还原酶之间充当一个远程且自由扩散的穿梭体角色。甲基紫精(MV2+)是常见的介体之一,但其毒性阻碍了它的广泛应用。目前,Cp*Rh络合物、天然黄素FMN等常作为替代电子传递介体使用[38-39]。
1.3 人工光促酶催化设计中的酶元件

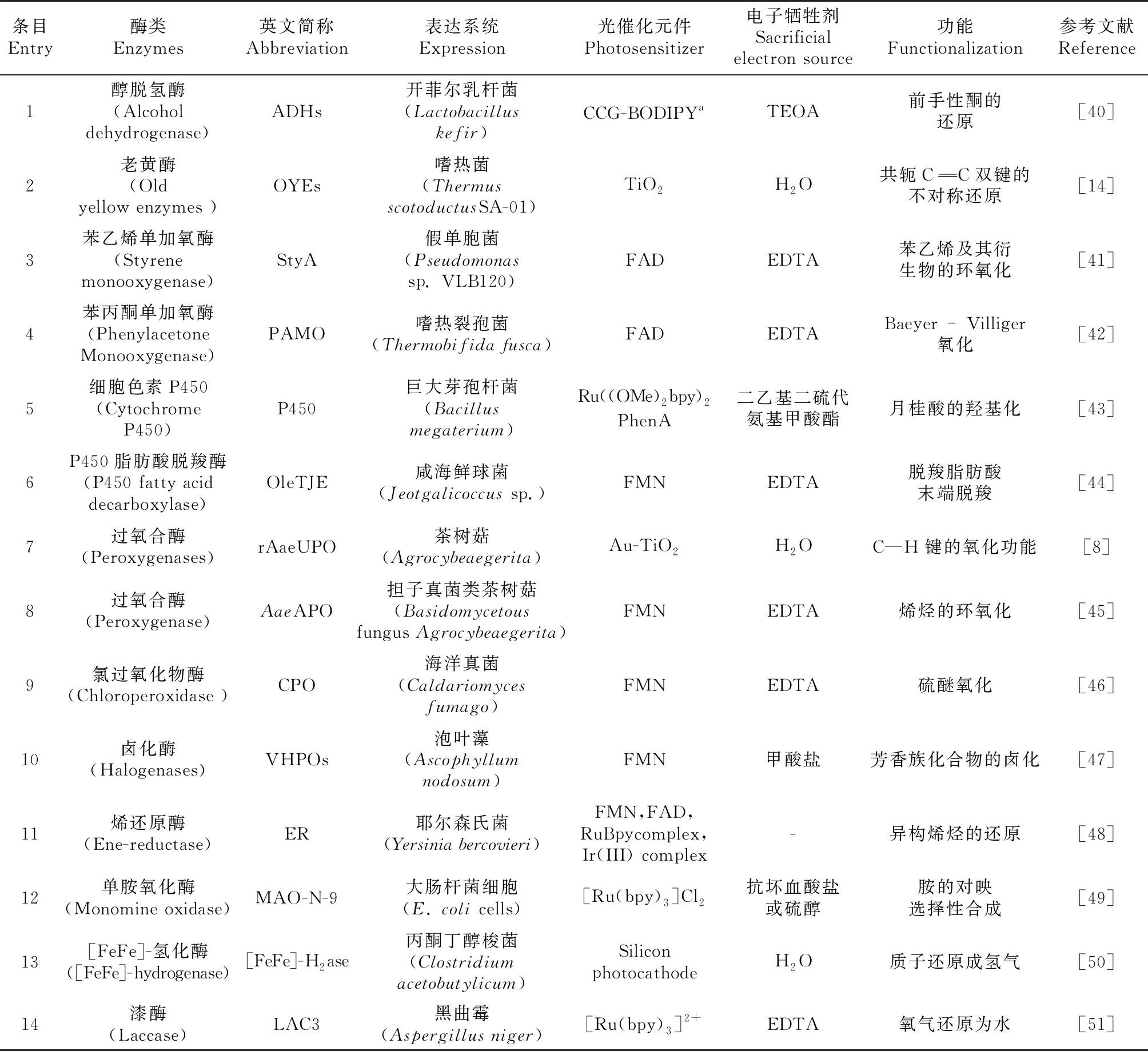
表1 光促酶级联反应中使用的酶类Table 1 Enzymes used in the cascaded photobiocatalysis
2 级联型光促酶催化体系的设计
目前,具有高活性和鲁棒性的级联型光促酶催化体系主要应用上述光催化元件和酶的不同组合。由于光催化反应通常在室温下进行,并可通过光激发电子和能量转移产生在水相中稳定且与生物酶兼容的中间体,这使级联型光促酶催化成为可能[16]。级联方式主要有3种:顺序、并发和协同(图 4)。顺序反应中酶催化和光催化反应条件不兼容,在进行多步反应时需要改变反应条件;而并发以及协同反应过程中,酶和光催化反应可以在同一条件下反应,通常采用“一锅法”的级联方式[2,48,52]。
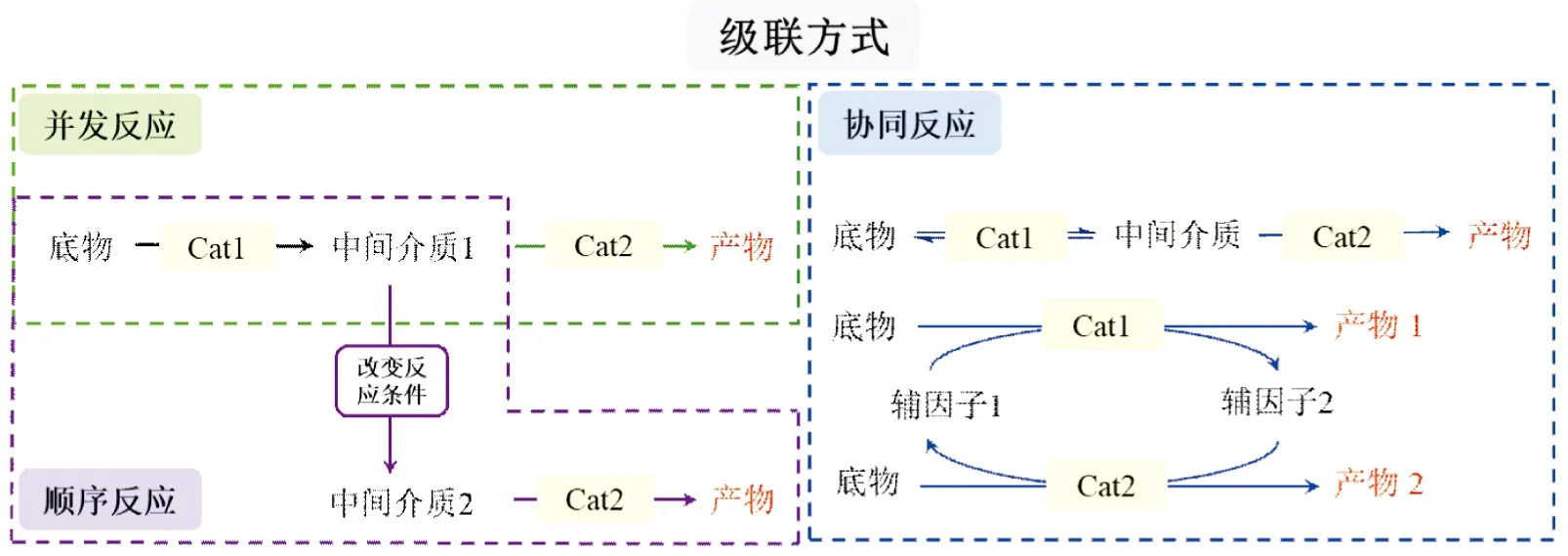
图4 级联型光促酶催化的3种反应方式Figure 4 Three pathways of cascading photocatalysis with enzymatic
在所有反应方式中,利用光催化元件为氧化还原酶提供氧化还原当量[NAD(P)H、FMN或者FAD]的研究最为广泛。例如,Choudhury等[40]设计了可见光驱动NAD(P)H再生策略来合成手性醇。在该方法中,以CCG-BO-DIPY为光催化元件,[Cp*Rh(bpy)Cl]Cl作为介体,采用“一锅法”进行光化学再生NAD(P)H来激活酮还原酶(ketoreductases,KREDs)还原多类型酮底物,获得了一系列手性醇产物,e.e.值达95%以上。同样,利用光催化元件辅助NADPH的循环,可为P450酶提供氧化还原当量,从而为P450酶选择性氧化反应提供了新思路。Lee等[18]实现可见光再生NAD(P)H来驱动胞外P450 BM3单加氧酶的脱烷基反应,通过使用曙红Y、电子牺牲供体TEOA及介体Cp*Rh(bpy)H2O实现NAD(P)H的再生。Mifsud等[14]使用Au掺杂的TiO2作为光催化剂,将水(同时作为反应溶剂)氧化产生的电子直接输送给OYE酶中还原型辅因子FMN,在可见光下反应6 h后,成功观察到茶香酮的还原(转化率为66%,e.e.为86%),具体见图5。
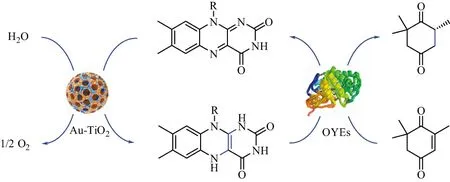
图5 Au-TiO2光催化剂与OYEs酶级联催化共轭CC双键的不对称还原Figure 5 Asymmetric reduction of conjugated CC bonds catalyzed by OYEs cascaded with Au-TiO2
H2O2作为一种还原态的氧供体,可以绕开氧化还原时复杂的电子传递链。过氧化物酶、水解酶、过氧合酶等均可使用H2O2作为共底物。但是,高浓度的H2O2会导致这类氧化酶的活性单元被氧化分解[6]。例如,从富马热酵母中提取的氯过氧化物酶(chloroperoxidase,CPO)在含有50 μmol/L H2O2的磷酸盐缓冲溶液(体积分数30%t-BuOH)体系中半衰期约为38 min[53]。可见,H2O2的浓度必须保持在最佳水平以维持H2O2依赖氧化酶的稳定性和反应活性。由于简单和廉价易得,葡萄糖氧化酶体系再生H2O2较为常用。但该方法的主要缺点(尤其是大规模应用)是较低的原子利用效率和反应中葡糖酸内酯(或相应的葡萄糖酸)的化学计量累积[54]。基于此,光催化原位生成H2O2的方法由于其操作稳定性高、底物转化率高等优势已成为一个重点研究方向。
Hollmann课题组设计了系列H2O2原位生成体系(图6)。利用黄素作为光催化元件,用电子供体(EDTA)对黄素进行光诱导,激发态的黄素与反应体系中存在的O2迅速自发反应生成H2O2,该体系可被应用于过氧合酶催化的羟基化、环氧化、磺氧化等反应[45];利用Au-TiO2无机光催化剂,创新性地提出并证实了理论上原子经济性100%的光催化H2O氧化原位生成H2O2,用于AaeUPO酶催化的C—H键选择性羟化反应[8];最近,水的光催化氧化原位生成H2O2进一步被拓展到辐射化学领域,放射性Co-60辐射或U-235(γ-射线)分解H2O并原位生成H2O2,后者可级联AaeUPO酶选择性催化乙苯生成(R)-苯乙醇等一系列氧官能化反应[55]。
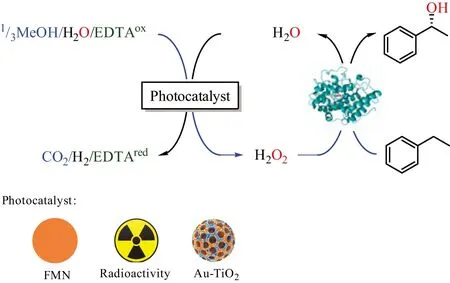
图6 3种光催化剂催化原位生成H2O2驱动AaeUPO酶Figure 6 Three photocatalysts used for n situ regeneration of H2O2 to drive AaeUPO
需要指出的是,H2O因其极为惰性(H2O的氧化能垒高达474.61 kJ/mol)导致氧化反应极其困难,使反应的转化率很低。而小分子醇如甲醇,在承担牺牲剂角色被彻底氧化的同时,也是出色的氢氧自由基捕获剂,可有效降低自由基对过氧合酶血红素的氧化。
上述级联光促酶催化可有效简化P450酶电子传递链,提升过氧化酶的稳定性。除此之外,可设计多种光催化反应和酶催化级联组合,即反应底物首先被光催化反应转化为反应中间体,后者作为酶的底物在接下来的过程中被直接转化。这种巧妙地结合简化了有机合成反应步骤,为不对称有机合成新反应途径的开发提供了很好的思路。
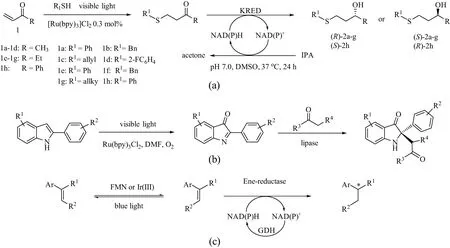
(a)合成1,3-巯基醇产物;(b)不对称合成2,2-双取代吲哚-3-酮;(c)CC键的不对称还原。图7 光催化反应与酶催化反应级联实例Figure 7 Examples of cascade photocatalytic and enzymatic reactions
Guo等[49]首次采用级联型光促酶催化方式设计了一种从环亚胺出发不对称合成R型环胺的循环网络反应体系。首先,使用水溶性铱催化剂[Na3Ir(sppy)3]将环亚胺还原为α-氨基烷基自由基,在抗坏血酸还原剂(AscH2)作用下,α-氨基烷基自由基通过HAT得到外消旋的环胺混合物。随后,通过单胺氧化酶(MAO-N-9)将S型环胺再氧化为环亚胺,如此进行循环。在连续光照射下,反应体系最终得到R型单一对映体。需注意的是,由于光催化反应和酶催化反应在细胞裂解液中相互干扰,MAO-N-9使用了大肠杆菌全细胞进行催化转化。最近,本课题组[58]采用水溶性的蒽醌催化剂,在缓冲液中顺利实现“一锅法”光促酶催化级联反应,可与多达11种酶级联合成光学纯的化合物。要指出的是,光诱导下产生的高活性氧和自由基会成为光促酶级联催化中抑制酶的主要因素,所以上述反应多采用“一锅两步法”。目前多数级联型光促酶催化反应以“一锅法”进行,无需进行中间产物分离,具有降低物耗和能耗、提高反应效率和绿色环保等特点。然而,由于光催化元件与酶通常在不同的介质和温度下工作,所以将酶与光催化剂以“一锅法”方式反应仍然具有挑战性。
3 诱导型光促酶催化体系的设计
第二种诱导型光促酶催化在反应过程中无需引入额外的光催化元件。目前已发现4类天然光促酶,即光合系统、光裂合酶、原叶绿素酸酯氧化还原酶以及脂肪酸光脱羧酶。光合作用系统作为地球上最重要的生物过程之一,由光系统I(PSI)和光系统II(PSII)共同促成了光合作用过程[59-60];光裂合酶是一种DNA修复酶,利用可见光修复被紫外线破坏的DNA[61-62];原叶绿素酸酯还原酶是一种依赖NADPH的光促酶,通过将原叶绿素转化为叶绿素a参与叶绿素的合成[63]。上述3类光促酶表明,光可以参与复杂的酶促反应。然而,上述酶在天然产物合成过程中的高度专一性限制了其在化学合成中的应用。在小球藻(ChlorellavariabilisNC64A)中提取纯化的第4类脂肪酸光脱羧酶(fatty acidphotodecarboxylase,CvFAP)则在化学合成中表现出巨大的潜力。FAP是一种蓝光-光促酶,可催化一系列游离脂肪酸的脱羧反应生成相应的烷烃和烯烃,且可以对体系反应时间进行精确控制[64](图8)。蓝光是诱导该酶活性中心FAD转变成激发态自由基并进行催化脱羧的唯一诱因。其中C16和C17脂肪酸的脱羧效率最高,转化率达到96%[65-66]。
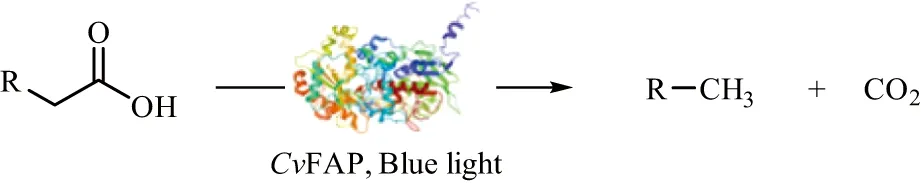
图8 CvFAP酶在蓝光下进行脱羧反应Figure 8 Decarboxylation catalyzed by CvFAP under blue light
为解决该光促酶只对长链脂肪酸具有活性的问题,可采用添加诱饵分子来提高对小分子羧酸的脱羧反应,实现H2、甲烷、烯烃等的制备,该策略可大幅度拓展光促酶的合成范围[67]。此外,与油酸水合酶级联(图9),可实现多种光学纯高级脂肪仲醇的合成,是替代传统格氏试剂法合成脂肪醇的一类有竞争力的绿色合成方法[68]。
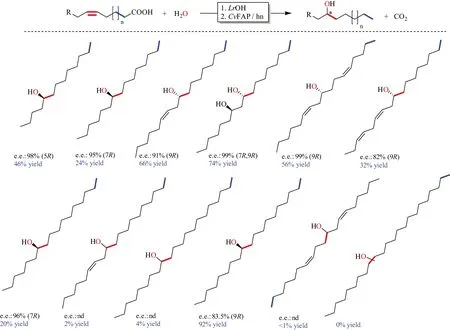
图9 CvFAP与LrOH酶级联催化不饱和脂肪酸合成手性脂肪仲醇Figure 9 Synthesis of chiral secondary fatty alcohols from unsaturated fatty acids catalyzed by cascading CvFAP and LrOH
如前所述,除自然界存在的光促酶外,许多经典的天然酶含有可对光响应的辅酶,如NAD(P)H、FAD、FMN等。在光激发下,辅酶形成还原性较强的自由基以改变原有的催化路径,实现新的催化过程。例如,NAD(P)H由于结构中含有1,4-二氢吡啶具有氧化还原活性,可被可见光直接激活使其从基态的弱单电子还原剂转变为强单电子还原剂,在光激发下还原一系列官能团[69]。Emmanuel等[15]发现,从罗尔斯顿菌和乳酸菌中提取出来的醇脱氢酶(RasADH和LKADH)在蓝光的激发下可作为自由基引发剂以及手性氢源(460 nm),将外消旋α-卤内酯不对称转化为脱卤内酯,转化率高达95%,产率为81%,e.e.值达到96%[图10(a)]。最近,该课题组又探索了黄素依赖的ER酶在光诱导下形成黄素自由基,实现了非天然条件下的立体选择性自由基环化反应。该反应以含烯基的α-氯酰胺作为底物,可选择性合成5-8元环内酰胺。其底物可包含多类型官能团,并可实现克级合成。鉴于内酰胺类化合物在药物合成中的突出作用,该反应具有重要价值[图10(b)][10]。以上两种光促酶催化反应都发生在分子内,而Huang等[17]最近设计了利用可见光直接诱导ER酶催化分子间的交叉偶联反应。通过使用端烯烃和α-卤化物羰基化合物实现自由基加氢-烷基化偶联反应,合成一系列γ-手性羰基化合物,具有良好的产率(高达99%)和对映选择性(e.e.值达到99%),突破了化学法催化的极限[图10(c)]。该反应中的关键步骤仍然是可见光诱导黄素自由基和底物自由基的形成。
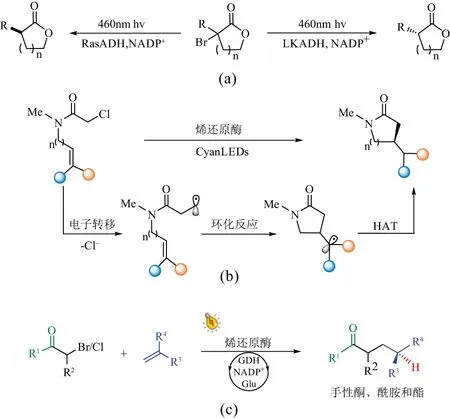
(a)光诱导醇脱氢酶进行脱卤内酯反应;(b)光诱导烯还原酶进行分子内环化反应;(c)光诱导烯还原酶进行分子间交叉偶联反应。图10 诱导型光促酶催化自由基的选择性生成及利用Figure 10 Selective generation and use of free radicals catalyzed by induced photobiocatalysis
4 光促酶催化在生物合成中的应用
20世纪末,生物催化的应用主要集中于合成或拆分光学活性中间体,并逐渐发展成为一种广泛适用于化学合成和制造的技术。但生物催化技术面临的一个长期挑战是酶功能开发不足。为此,酶源挖掘及酶工程等已成为突破上述瓶颈的手段。而光促酶催化技术已被证明是一种开发酶功能的新途径。除了将光催化和酶催化反应进行级联组合开发新的生物催化途径外,也可通过结合酶的定向进化改造,有力地促进天然酶的非天然催化范围,在新分子合成和新化学开发方面具有重要潜力。浙江大学吴起等[70]利用迭代饱和突变,对脂肪酸光促脱羧酶的残基Y466与核黄素光催化单元周围的关键区域氨基酸位点进行改造,成功实现了α-羟化脂肪酸和少数氨基酸的动力学拆分,获得的光学纯羟化脂肪酸可作为砌块用于抗癌药或神经中枢抑制器的合成。采用类似的策略,改造后的光促脱羧酶可催化动力学拆分外消旋草铵膦,首次实现了酶催化重要农药L-草铵膦的克级制备[71]。而上述拆分反应在自然酶催化过程活性极低或无法发生。可见,光促酶催化在生物合成中极具开发潜力。表2列举了近几年一些典型的人工设计的光促酶级联催化应用实例,以便为手性及区域选择性的化学合成提供参考。
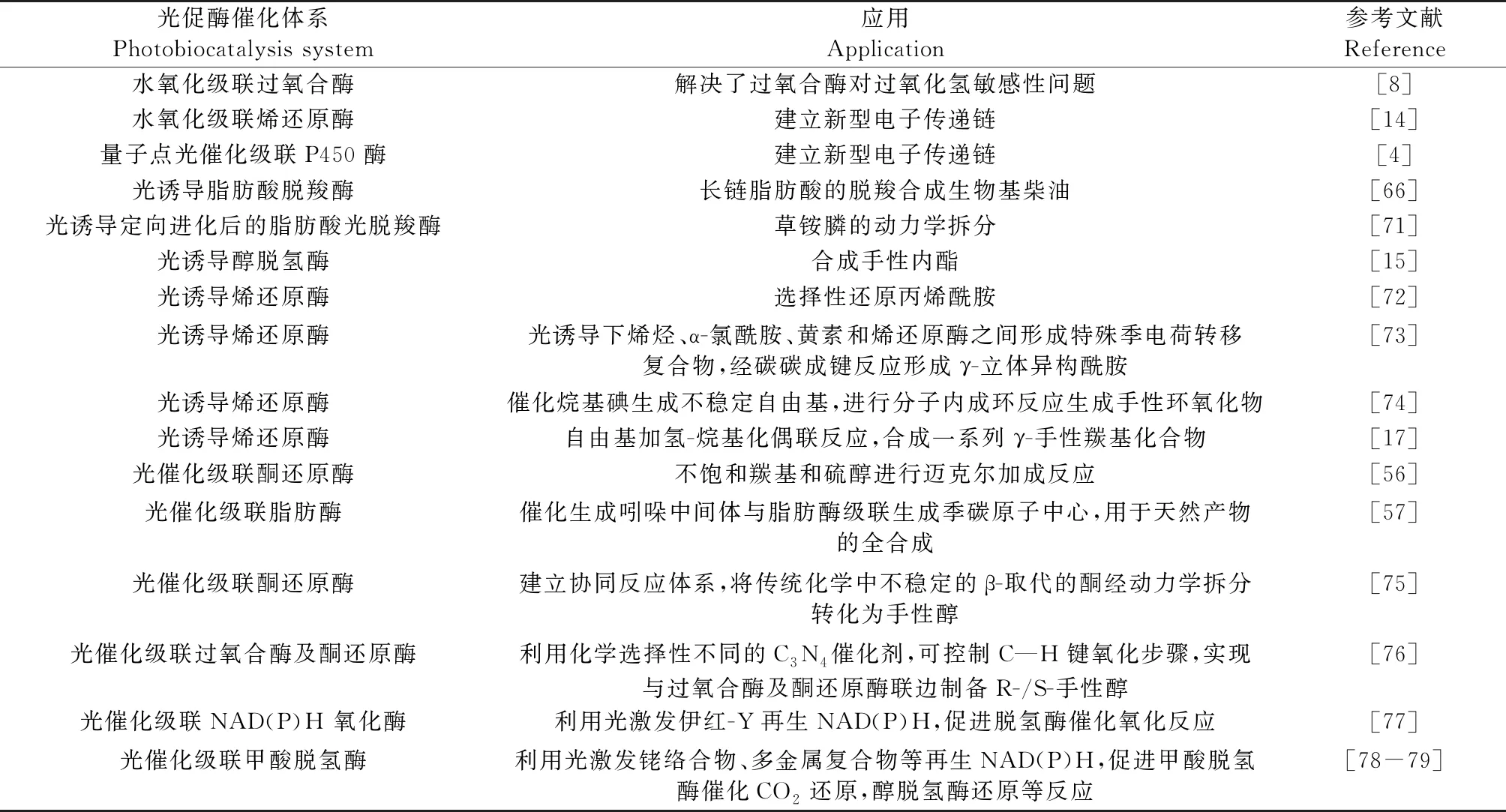
表2 光促酶催化在生物合成中的应用实例Table 2 Examples using photobiocatalysis in biosynthesis
5 小结与展望
光促酶催化是一种迅速发展的多学科交叉生物合成技术,在新分子合成及新化学开发方面显示出广阔的前景。但是,在光促酶催化领域面临多个亟待解决的问题。首先,在第一类级联型光促酶催化方式中,反应体系多是简单地将光敏剂、电子媒介、辅酶等与酶催化剂按照一定的配比组合。在该情况下,激发电子的随机传递和转移将显著影响光能的利用效率。因此,可设计光催化元件与酶元件的集成体系,构建有效的光-酶催化界面以更高效地促进电子在两个催化元件间的传递。其次,光催化元件的催化活性是影响光促酶催化效率的限制性因素。因此,不断地改进或设计新型的高活性光催化元件对提升光促酶催化的效率和应用范围至关重要。对光催化装置的设计优化以及对酶元件与光催化元件的共固定,均可显著提高反应的转化率和酶的稳定性。最后,近年围绕烯还原酶的设计改造工作虽然已充分展示了酶改造的强大作用,但是在第二类诱导型光促酶催化方式中,反应多在无氧条件下进行,其操作复杂性在一定程度上限制了其在合成化学中的潜力。因此,酶定向进化技术及结合机器学习的酶智能设计与改造方法有望极大地推动光促酶催化技术的发展[80-84]。未来的酶定向进化不仅要开发光促酶的新功能,提升光促酶的催化效率、区域及立体选择性、热稳定性、底物普适性等,还要致力于获取无需在隔绝氧气条件下(如改变酶活性孔道与氧气的结合能力)可进行催化反应的人工酶。
综上所述,随着上述挑战的解决,光促酶催化技术在化工、医药、农药等合成领域的应用将会不断拓展,这必将有力促进不对称催化合成和绿色生物制造领域的高质量发展。
