靶向小鼠Tyr基因的CRISPR/Cas9系统构建及打靶效力分析
2021-10-25岳鹏鹏杨光宇王添贤肖义然于鸿浩
岳鹏鹏,杨光宇,王添贤,付 灿,肖义然,于鸿浩
(1.桂林医学院 生物技术学院,桂林 541199;2.桂林医学院 公共卫生学院,桂林 541199;3.北京体育大学 运动人体科学学院,北京 100084)
CRISPR/Cas9系统发挥基因打靶功能时,Cas9核酸内切酶在向导RNA(small guide RNA,sgRNA)的介导下结合到基因组目标靶点位置,切割DNA,最终达到基因编辑的目的[1]。Cas9核酸内切酶结合靶点的关键是sgRNA。因此,能够设计高效靶向目标基因的sgRNA是CRISPR-Cas9系统能否特异性编辑目标基因的先决条件。在基于CRISPR/Cas9系统敲除基因、探讨基因功能的研究中,sgRNA通常选择目标基因前1/2外显子作为设计区域;涉及多个转录本的基因时选择保守的外显子区域设计靶点;同时利用在线软件评估其脱靶效应,选择低脱靶效应的位点作为靶点。其目的是在靶点位置形成移码突变,导致基因功能失活,进而研究基因功能[2]。然而,在一些特殊目的的研究中,sgRNA的设计具有其特殊性。如农业研究中,FecB基因的p.Q249R突变被证明是与羊的多胎性状有关,为了制备该基因突变的绵羊,则必须在p.Q249R位点附近设计sgRNA[3]。同样,医学遗传学研究中,通常根据发现的变异位点创新性设计sgRNA,以研究基因变异的致病机制或探讨治疗手段等[4]。
人类白化病是以黑色素缺乏或缺失为主要临床表现的疾病。根据色素缺失的受累部位可将其分为两类,即眼皮肤白化病(oculocutaneous albinism,OCA)(色素减少或缺失涉及皮肤、毛发、眼睛)和仅限于眼睛的眼白化病(ocular albinism,OA)[5]。我国白化病平均发病率为1∶17 000[6],至少存在OCA1(I型)、OCA2和OCA4共3种类型[7],其中I型最为普遍[8],也是临床表现及危害最为严重的类型[9]。酪氨酸酶 (tyrosinase,TYR)基因突变是引起I型白化病的主要病因[10]。但TYR基因突变具有多种类型,包括点突变、无义突变以及错义突变等,不同的突变可能引起的临床症状不同,患病程度亦不相同。因此,有必要筛查明确的、典型的TYR致病突变位点,并将其定位于小鼠基因组上,然后通过CRISPR/Cas9系统对该位点进行打靶,建立高效编辑系统,从而为建立可模拟人类白化病的小鼠动物模型奠定基础。
1 材料与方法
1.1 材料
1.1.1 质粒与细胞
实验所用的CRISPR/Cas9系统包括两种质粒,分别为sgRNA表达质粒pGL3-U6-sgRNA-PGK-puromycin(addgene #51133)和Cas9表达质粒pST1374-NLS-flag-linker-Cas9(addgene #44758),均由上海科技大学黄行许教授惠赠。小鼠N2a细胞由广州大学周建奎博士惠赠。
1.1.2 主要试剂与仪器
大肠杆菌感受态菌株DH5α和pMDTM19-T Vector Cloning Kit购自宝日医生物技术(北京)有限公司;去内毒素质粒中提试剂盒购自北京康为世纪生物科技有限公司;BsaI购自NEB公司;胎牛血清、DMEM培养基、LipofectamineTM 3000 Transfection Reagent(InvitrogenTM)试剂盒、杀稻瘟霉素、嘌呤霉素均购自Thermo Fisher Scientific公司;TransDirect®Mouse Genotyping Kit购自北京全式金生物技术有限公司;Solution I、dNTP Mixture、TaKaRa ExTaq和10×ExTaqBuffer、PremixTaq酶均购自宝日医生物技术(北京)有限公司;DNA纯化回收试剂盒购自天根生化科技有限公司;引物由上海百力格生物技术有限公司合成;测序由广州生工生物工程股份有限公司完成。实验所需的主要仪器包括超净工作台、CO2培养箱、台式离心机、PCR仪、核酸浓度测定仪、低温冷冻离心机、摇床和超低温冰箱等。
1.2 方法
1.2.1 人TYR基因致病突变位点的筛查
OMIM,即“在线人类孟德尔遗传”数据库,收录了遗传病、性状和基因的相关信息。登录该数据库,检索导致白化病的TYR致病突变。鉴于OMIM收录的突变位点有限,本研究亦在ExAC(外显子组整合数据库)检索TYR基因的变异情况,并进行变异频率排序分析,为sgRNA的设计作为参考。最后,为了确定TYR基因变异位点的致病性,本研究继续在ClinVar数据库筛查TYR突变的致病情况,排除非致病性、可能致病性的变异位点。综合分析以上3个数据的结果,从而找到明确的、典型的强致病位点。
1.2.2 sgRNA的设计及表达载体的构建
由于人和小鼠的物种差异,利用Clustal Omega软件比对了人TYR和小鼠TYR蛋白序列,以找到与人致病位点相对应的氨基酸定位,即小鼠拟突变位点。然后从Ensembl数据库中导出小鼠Tyr基因序列,找到拟突变位点的编码序列。利用Vector NTI软件的“Find Motifs”功能在拟突变位点编码序列附近搜索具有5′-N(21)GG特征的位点,设计了3个sgRNA,分别为Tyr-M-sg1、Tyr-M-sg2和Tyr-M-sg3。
在3个sgRNA序列的5′端加上“accg”、互补链5′端加上“aaac”合成得到正反向寡核苷酸,经TouchDown程序退火,形成具有黏性末端的双链DNA片段。
利用BsaI限制性内切酶酶切pGL3-U6-sgRNA质粒,得到酶切产物pGL3-U6-sgRNA-BsaI。然后琼脂糖凝胶电泳检测酶切结果并回收,得到pGL3-U6-sgRNA-BsaI质粒用于后续连接实验。
将具有黏性末端的双链DNA片段和酶切产物pGL3-U6-sgRNA-Bsa I进行连接。连接产物转化大肠杆菌,37 ℃过夜培养20 h后,挑取单克隆并用通用引物U6测序,鉴定出阳性克隆,于37 ℃、220 r/min对阳性克隆摇菌,去内毒素中提试剂盒提取质粒,分别得到pGL3-U6-Tyr-sgRNA1、pGL3-U6-Tyr-sgRNA2和pGL3-U6-Tyr-sgRNA3质粒。
1.2.3 细胞转染及药物筛选
将小鼠N2a细胞培养于含10%胎牛血清的DMEM完全培养基中,于37 ℃,5% CO2浓度,饱和湿度的培养箱中培养,每2~3 d更换新鲜培养基,待细胞汇合度达到80%~90%后,以0.25%胰蛋白酶消化,传代接种至6孔板中,16~18 h后,待细胞汇合度达到60%~70%时,进行转染。
转染采用LipofectamineTM 3000 Transfection Reagent(InvitrogenTM)试剂盒分别将pGL3-U6-Tyr-sgRNA1质粒、pGL3-U6-Tyr-sgRNA2质粒、pGL3-U6-Tyr-sgRNA3质粒,以及pST1374-NLS-flag-linker-Cas9质粒,共转染小鼠N2a细胞,然后利用嘌呤霉素和杀稻瘟菌素进行药物筛选,以获得阳性共转染细胞。
1.2.4 细胞裂解及PCR扩增
根据TransDirect®Mouse Genotyping Kit的操作说明,对实验步骤进行了少许调整。将收集的3组阳性共转染细胞加入8 μL的AD1悬浮细胞沉淀,然后取8 μL液体至PCR管,加入2 μL的AD2,55 ℃孵育10 min,95 ℃ 3 min;加入8 μL的AD3混匀,分别得到3组细胞的细胞裂解液,作为后续打靶位点DNA的PCR扩增的模板,-20 ℃保存。
分别以上述3组细胞的细胞裂解液为模板,进行打靶位点DNA的PCR扩增反应,引物序列见表1。3组打靶位点的PCR扩增反应体系:细胞裂解液2 μL,上游引物1 μL,下游引物1 μL,dNTP Mixture 2 μL,TaKaRa ExTaq1 μL,10×ExTaqBuffer 2.5 μL,灭菌水补充至25 μL。反应的程序:95 ℃预变性5 min;95 ℃变性20 s,72 ℃退火20 s,-1 ℃/循环(即每循环1次降低1 ℃),72 ℃延伸25 s,共10个循环。95 ℃变性20 s,62 ℃退火20 s,72 ℃延伸25 s,共25个循环;72 ℃5 min,16 ℃∞。将上述3组PCR产物进行Sanger测序。如果PCR产物打靶位点出现套峰,则初步确认发生了基因编辑。剩余PCR产物-20 ℃保存,用于后续的TA克隆实验。

表1 引物序列Table 1 Primer sequences
1.2.5 TA克隆测序分析
将3组PCR产物进行胶回收,利用Solution I 将纯化后的DNA与pMDTM19-T载体进行连接。将连接产物转化至感受态大肠杆菌中,涂布LB固体培养基上培养20 h后,进行菌落PCR反应挑选阳性克隆。其菌落PCR反应体系:菌落水溶液1 μL,PremixTaq5 μL,上游引物0.5 μL,下游引物0.5 μL,灭菌水补充至10 μL。上下游引物分别为通用引物M13F和M13R。菌落PCR的反应程序:95 ℃预变性5 min;95 ℃变性20 s,60 ℃退火20 s,72 ℃延伸25 s,26个循环;72 ℃再延伸5 min。反应结束后电泳鉴定。将筛选出的阳性克隆进行Sanger测序,测序结果与野生型序列进行比对分析。
2 结果与分析
2.1 人TYR致病位点筛查结果
利用OMIM在线数据库筛查到I型白化病是由TYR基因突变引起的,包括38种致病突变(表2);ExAC数据库共筛查到283个突变位点(表3);利用ClinVar数据库检索TYR致病位点的突变情况,共检索到65个突变位点(表4)。根据3个数据库的综合结果最终确定人TYR基因第81号位的脯氨酸(简称Pro)位点为拟突变位点。

表2 OMIM数据库中人TYR基因的致病突变情况Table 2 Pathogenic mutation of human TYR gene in OMIM database
表3 ExAC数据库中人TYR基因突变情况Table 3 Human TYR gene mutation in ExAC database

表4 ClinVar数据库中人TYR基因致病突变位点情况Table 4 Pathogenic mutation sites of human TYR gene in ClinVar database
2.2 sgRNA设计及载体构建结果
蛋白序列比对分析发现人TYR基因第81号位的Pro与小鼠Tyr基因第81号位的Pro相对应(图1),即小鼠Tyr第81号位的Pro编码序列为拟突变位点。根据小鼠编码序列设计了3个打靶位点(图2)。通过DNA合成(表5)、退火、连接转化以及测序等步骤,成功构建了3个位点的sgRNA表达载体。

黑色箭头为人和小鼠81号位Pro氨基酸。图1 人TYR和小鼠Tyr的蛋白序列Figure 1 Human TYR and mouse Tyr protein sequences
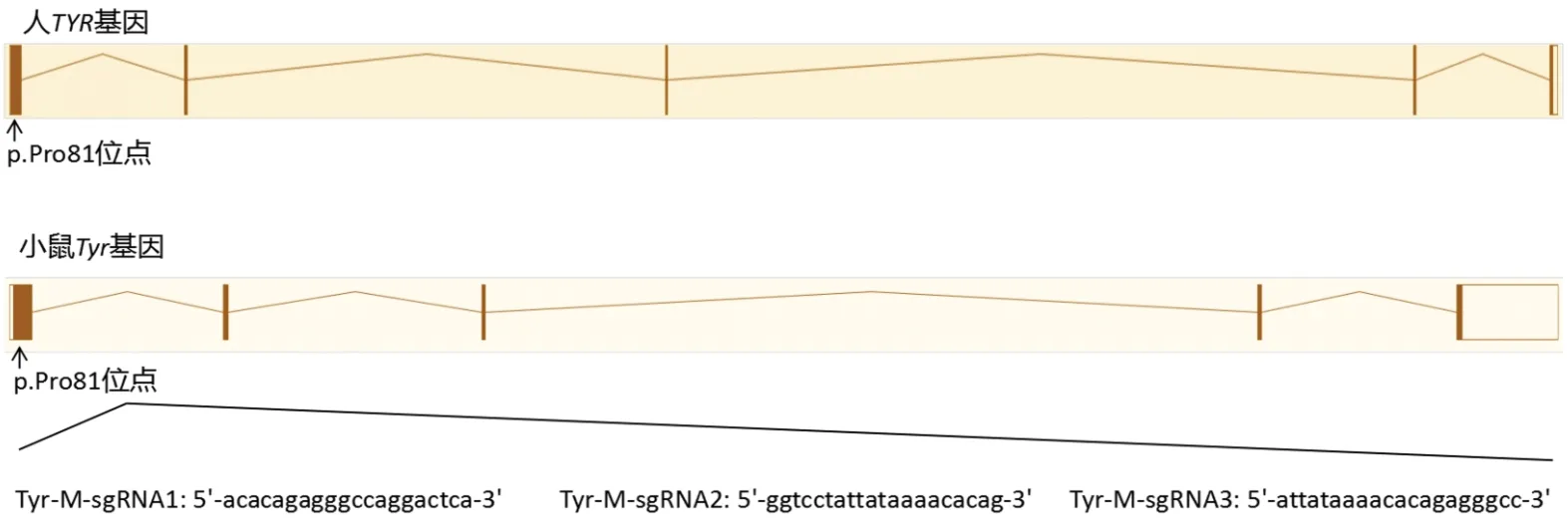
图2 小鼠Tyr基因打靶位点的设计图Figure 2 Design map of mouse Tyr gene target site

表5 化学合成的sgRNA序列Table 5 Chemically synthesized sgRNA sequences
2.3 细胞转染及药物筛选结果
转染时六孔板的每个孔转染sgRNA表达质粒和Cas9表达质粒均为2.5 μg。转染24 h后,用浓度为50 μg/mL嘌呤霉素和浓度为100 μg/mL的杀稻瘟菌素进行药物筛选48~62 h,得到阳性的共转染细胞(图3)。然后,将阳性共转染细胞用磷酸盐缓冲液洗涤3次,用0.25%的胰蛋白酶消化,并离心收集,用于后续实验。
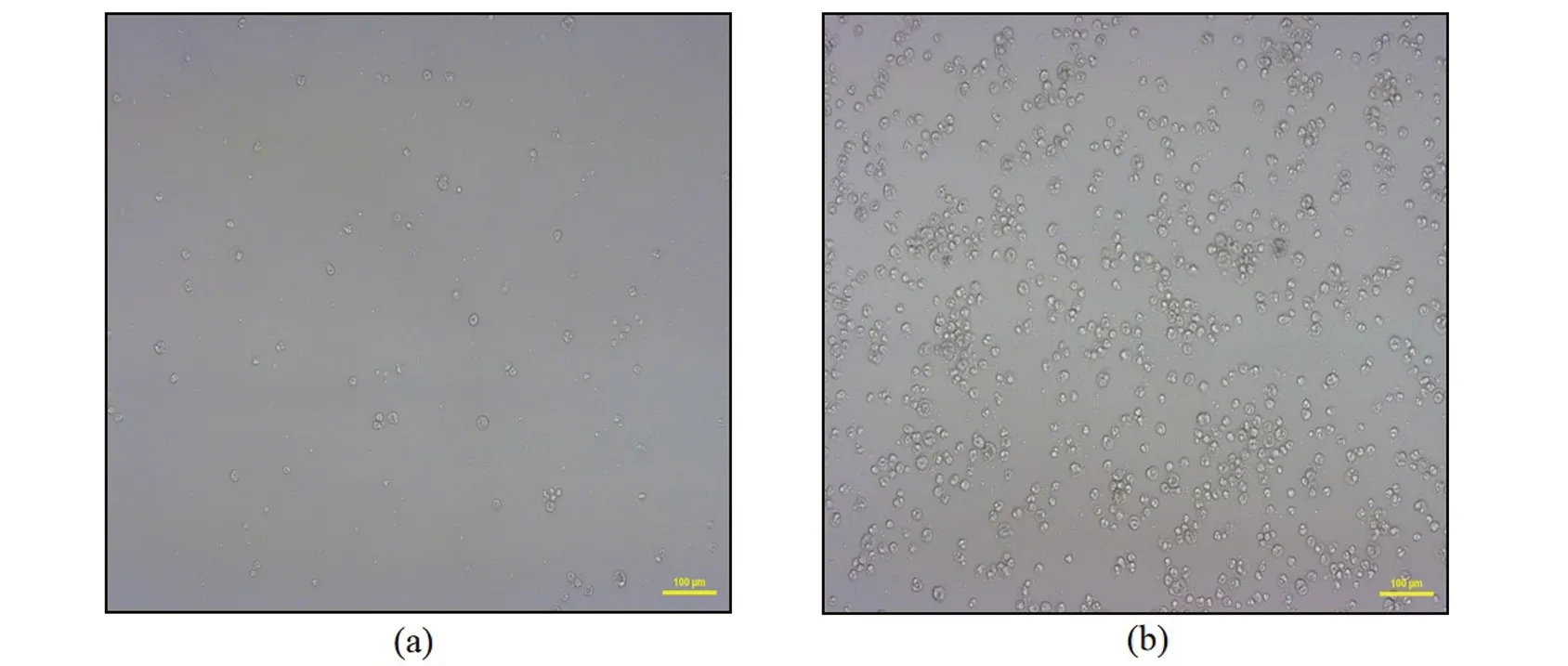
(a)对照组细胞;(b)pGL3-U6-Tyr-sgRNA2和pST1374-NLS-flag-linker-Cas9共转染药筛阳性细胞。图3 阳性共转染细胞Figure 3 Positive co-transfected cells
2.4 打靶区域PCR产物测序及TA克隆测序结果
收集的阳性细胞经过裂解和PCR扩增,得到了包含打靶区域的DNA片段,电泳检测后,进行测序。测序结果发现在3个靶点位置均有套峰出现,表明发生了DNA序列的改变,基因打靶成功(图4)。
2.5 TA克隆测序结果
打靶成功的PCR产物经过胶回收、连接转化、菌液PCR鉴定以及阳性克隆测序等步骤,得到了基因编辑后的具体序列。测序结果与野生型序列比对分析表明,在sgRNA1、sgRNA2和sgRNA3靶点位置发生了碱基的随机插入和缺失(图5),编辑效率均为100%。
3 讨论
CRISPR/Cas9系统被称为第三代基因编辑系统,与应用较早的基因编辑技术如锌指蛋白核酸酶(ZFN)[11]或转录激活因子样效应物核酸酶(TALENs)[12]相比,具有设计灵活、操作简便、准确性高、成本较低、可同时多位点打靶等优势。本研究构建了靶向小鼠Tyr基因的高效CRISPR/Cas9系统,TA克隆测序分析表明所设计的3个靶点均达到了100%的打靶效率。如此高的打靶效率首先取决于sgRNA的设计,同时还与转染效率等试验细节密切相关。如本研究中使用了Lipofectamine TM 3000 Transfection Reagent (InvitrogenTM)试剂盒,转染效率明显好于Lipofectamine TM 2000转染试剂;实验中严格控制细胞的生长状态、细胞接种密度、转染时间以及药物筛选时间等实验要素;使用全式金TransDirect® Mouse Genotyping Kit,其独特裂解液裂解细胞后无需纯化,裂解物可直接作为模板进行后续扩增,提高了实验效率。当然100%的编辑效率是采用TA克隆方法得到的结果,由于受到测序样本量的限制其结果的精准程度不如高通量测序高。
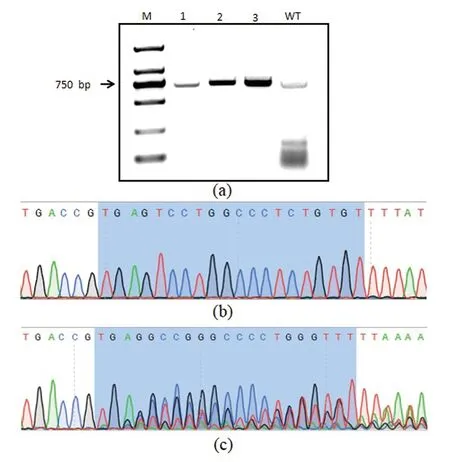
(a)打靶位点PCR产物电泳图[M:DNA ladder(DL2000);1、2和3分别代表包含sgRNA1、sgRNA2和sgRNA3打靶位点的PCR产物;WT为野生型PCR产物]。(b)野生型sgRNA1打靶位点测序峰图(深色背景部分为sgRNA1序列)。(c)基因编辑sgRNA1打靶位点测序套峰图(深色背景部分为DNA序列发生改变的sgRNA1序列)。图4 PCR产物电泳及测序图Figure 4 PCR product electrophoresis and sequencing diagram

加粗字体为打靶序列;斜体字体为PAM结构;“-”代表缺失碱基;小写字母代表插入碱基;“……”代表未列出序列;“-N”代表缺失碱基数量;“+N”代表插入碱基数量;“N/N”代表基因型数量及阳性克隆测序总数。图5 TA克隆测序结果Figure 5 TA clone sequencing results
TYR基因由5个外显子组成,其基因组DNA约为65 kb,编码由529个氨基酸组成的TYR。TYR是一种含铜的酶,催化黑色素生物合成途径的前两个步骤,将酪氨酸转化为L-二羟基苯丙氨酸(DOPA),然后转化为DOPA多巴醌[13]。该蛋白是催化黑色素生成的关键酶类之一,若其发生突变将导致不同程度的皮肤、头发和眼睛的色素沉着减少,从而引起人类白化病[14-15]。郑辉等[16]利用生物信息学软件预测了11种TYR基因变异后的蛋白构象变化,发现11种变异导致蛋白质构象变化各不相同,揭示了突变的致病性与蛋白结构和功能的改变相关。推测TYR突变导致蛋白质构象的差异可能是白化病临床症状异质性的主要原因。目前,研究者以Tyr基因为靶点,基于CRISPR/Cas9以及碱基编辑器等工具制备了Tyr基因编辑小鼠[17-19]、兔子[20]、斑马鱼[21]和猪[22]等,以上基因工程动物均检测到白化表型,表明以通过TYR基因工程动物模拟人类的白化病是可行的。本文与上述研究相比最大的特点是通过检索数据库,仔细分析明确的、典型的TYR强致病突变位点,并将其定位于小鼠基因组上,为后续建立Tyr基因编辑小鼠提供了准确的靶位点信息。总之,我们设计并验证了行之有效的靶向小鼠Tyr基因的CRISPR/Cas9系统,满足TYR基因编辑小鼠制备的需要。
4 结论
本研究通过筛查白化病致病基因TYR的变异情况,明确了高致病性的突变位点,并将其定位于小鼠基因组上。然后基于CRISPR/Cas9系统设计了3个靶点,并在小鼠N2a细胞上实现了100%效率的基因打靶,成功构建了模拟人高致病突变位点的靶向小鼠的基因编辑系统,为后续建立基因工程动物模型奠定基础。
