Single-cell omics analyses with single molecular detection:challenges and perspectives
2021-10-19GradimirMisevic
Gradimir Misevic
Research and Development, Gimmune GmbH, 6302 Zug, Switzerland.
Abstract The ultimate goal of single-cell analyses is to obtain the biomolecular content for each cell in unicellular and multicellular organisms at different points of their life cycle under variable environmental conditions. These require an assessment of: a) the total number of cells, b) the total number of cell types, and c) the complete and quantitative single molecular detection and identification for all classes of biopolymers, and organic and inorganic compounds, in each individual cell. For proteins, glycans, lipids, and metabolites, whose sequences cannot be amplified by copying as in the case of nucleic acids, the detection limit by mass spectrometry is about 105 molecules. Therefore, proteomic, glycomic, lipidomic, and metabolomic analyses do not yet permit the assembly of the complete single-cell omes. The construction of novel nanoelectrophoretic arrays and nano in microarrays on a single 1-cm-diameter chip has shown proof of concept for a high throughput platform for parallel processing of thousands of individual cells. Combined with dynamic secondary ion mass spectrometry, with 3D scanning capability and lateral resolution of 50 nm, the sensitivity of single molecular quantification and identification for all classes of biomolecules could be reached. Further development and routine application of such technological and instrumentation solution would allow assembly of complete omes with a quantitative assessment of structural and functional cellular diversity at the molecular level.
Keywords: single-cell analyses, single molecular detection, ome, transcriptomics, proteomics, glycomics,lipidomics, metabolomics
Introduction
Single-cell analyses are the complex and multidisciplinary set of studies that employ the fundamental knowledge of biology, medicine, chemistry, physics,informatics, and their accompanying technologies.This new research field is emerging from classical omics performed on populations of thousands to millions of cells due to a relatively low detection sensitivity for biomolecules. The resulting data described average cell omes. In order to obtain information about the existence and the nature of cellular heterogeneity and its relation to stochastic,genetic, and environmental factors, many of which may be hidden in the average ome, single-cell omics aim to increase the sensitivity of measurements to the single molecular level, and achieve high throughput processing of millions of cells.
The term "ome" is the suffix abstracted from ancient Greek that is used in life sciences to refer to some sort of totality. Therefore, the suffix "ome" is addressing the object of the study. According to omics organization[1], the ome and omics are defined as follows: "Ome means the organic totality of something, some category, or some action"; "Omics is a general term for a broad discipline of science and engineering for analyzing the interactions of biological information of objects in various "omes".
Ome and omics have been hierarchically categorized into biological and nonbiological objects. The biological ome is therefore named biolome and would be referred to as omics of bio-objects. According to the biological object or biological process, biolome is further subclassified by omics organization into 11 categories: 1) system, 2) molecule, 3) structure,4) function, 5) process, 6) anatomy, 7) method,8) evolution, 9) biodiversity, 10) environment, and 11) social. Although many of these categories can be sorted using different approaches and rules, the importance is that any ome and omics must be precisely defined according to the tight connections and overlapping of the study objects.
At the cellular level, ome is named cellome, which provides quantitative information about the total number of cells and the number of different cell types in the selected studied organism,e.g., the human body(human cellome). The analyses required to obtain the complete cellome are termed cellomics which would quantify the total number of all cell types in a particular organism and/or in the organ at the certain developmental and life cycle stage, as well as in the specific disease. Cellome shall also contain data about allogeneic variation within the study species.Therefore, correct labeling of the study object must be added to cellome,e.g., human cellome, or more narrowed object,e.g., human liver cellome, or human liver cancer cellome.
Similarly, at the molecular level, the ome can be either any biopolymer type, or organic molecule, or inorganic compound, within a particular organism and/or a particular cell type at a particular stage of the organism's life cycle. The omics would be the analytic process of acquiring quantitative and complete information about the specific molecule. Therefore,the research field of omics analyses, when considering each type of molecule, can be further divided into an extremely large number of subdisciplines[1]. Common subdisciplines based on the type of biological molecules studied are genomics for obtaining genome,transcriptomics for transcriptome, proteomics for proteome, lipidomics for lipidome, glycomics for glycome, and metabolomics for metabolome. The systems that these subdisciplines are studying are usually species individual organisms; specific populations of organisms based on age, place and/or condition of living, and/or stage of disease; organs and tissues types; and cell types.
Single-cell omics is dedicated to complete analyses of all individual cells in tissues, organs, organisms, or population of organisms, and not on populations of cells. Therefore, quantification and identification of all molecules with single molecular detection is the only way towards the assembly of the whole ome that contains the complete molecular catalog of each individual cell in a study object.
The first part of this review provides a comprehensive list of single-cell omics challenges related to the quantitative measurement of molecular content of individual cells. Following is the overview of the present analytics limits and perspectives for high throughput micro and nanotechnological solutions.The progress in this field could lead to the identification and quantification of all classes of biomolecules with single molecular detection sensitivity in each individual cell, and thus reveal the molecular basis of cellular diversity.
Challenges of single-cell analyses with quantitative single molecular detection and identification
Single-cell omic analyses with single molecular detection are facing four tightly interconnected and extremely challenging goals.
1. To obtain the total number of cells and degree of their diversity in individual multicellular organisms during: a) different developmental stages, and b) their adult life cycles.
2. To apply the single molecular detection, quantification, and identification for all cellular: a) biopolymers, b) organic compounds, and c) inorganic compounds.
3. To build the quantitative catalogs of: a) the complete molecular content in each individual cell,b) the spatiotemporal subcellular organization of all molecules, and c) changes in the molecular content and their subcellular localization associated with healthy circadian physiological variations, with diverse pathological cases, with therapeutic treatments, all under variable environmental conditions.
4. To develop technologies and instrumentation for:a) non-destructive single cell isolation from all tissues and organ types, b) high throughput systems for parallel processing of individual cells, c) measurements and analyses of the complete molecular content by ultrasensitive quantification of all biopolymer,organic and inorganic compounds with the single molecule identification and quantitative detection sensitivity in each analyzed cell, and d) bioinformatic tools for collecting, analyzing, and storing data.
Reaching these highly demanding goals of quantifying the total number of all molecule types in each of all the identified cells in an individual organism will result in the ultimate structural and functional ome fingerprint. Due to the lack of highly developed micro and nanotechnological solutions, this is at the present not achievable for a higher level of multicellular organism contains trillions of cells, with each cell comprising of about 1 to 10 billion molecules[2–4]. Furthermore, it would be necessary to measure xenogeneic, allogenic, developmental, and environmentally dependent changes, which adds another three layers of complexity in measurements:handling, analyzing, and storing the enormous amount of collected data.
Workflow of single-cell analyses
Single-cell omics, which involves the detection and identification of all molecules in each individual cell,uses two general approaches. The first one is based on biochemical analytics, which is requiring mechanical destruction and solubilization of cell structure, in old jargon known as "smash and spin", followed by separation, quantification, and identification of all classes of biomolecules. The second approach for single-cell analyses is nondestructivein vivomicroscopy. It is examining the expression level of individual color-coded tagged molecules and their subcellular localization by super-resolution optical microscopy.This approach offers the possibility to study simultaneously many living cells during their life cycle and to detect and follow precisely the position and fate of a single labeled molecule in space and in time[5–9].However, only a few labeled molecules in the transparent organisms, or in cell culture are possible to observe. This approach is providing excellent comparative results for a few selected molecules in different cells that can be studied under different conditions,but obviously, it does not result in the assembly of the complete cellular ome. The related, but destructive,single cell imaging is based on matrix-assisted time of flight mass spectrometry[10], or time of flight secondary ion mass spectrometry (SIMS) with the capability of 3D scanning[9,11]. They have limited sub-micrometer resolution and relatively low sensitivity, which is unable to perform the single molecule detection.
Depending on the nature and the origin of the selected specimen, objectives of the study, and sensitivity of the particular set of methodologies to be used, the results obtained will lead to the assembly of either complete or partial omes. Since analyses of cells require the necessary step of solubilization at a specific time point, the fate of each individual cell cannot be tracked by an analytic cell destructive approach as in the live cell imaging. Heterogeneity of cells in a population may be the consequence of either physiologically, and/or stochastically induced changes,involving also cell cycle asynchrony. Therefore, in order to unveil causalities underlying the origin of cellular heterogeneity, single-cell omics requires a combinatorial approach of destructive analytics and nondestructive live imaging that can follow dynamics of expression and localization of biomolecules.
The workflow of single-cell analyses consists of the following five methodological steps that are based on the complex set of technologically challenging procedures, many of which need to be improved.
Minimally invasive preparation of single cells from the examined samples (tissue, organ, organism)
This step is particularly difficult when handling solid tissues that need mechanical micromanipulation and/or enzymatic treatment for cell dissociation. Such procedures alter physiological states of cells that may cause changes in transcriptome, proteome, glycome,lipidome, and metabolome. Furthermore, these alterations may differ among individual cells present in the studied cell population. Contrary, blood cells are readily available as single cells in solution without any treatment.
Cell separation
It requires high throughput instrumentation to simultaneously process hundreds to thousands of individual cells. Either flow cytometry equipped with cell sorting, or microfluidic devices, or automated parallel micro pipetting can be used.
Single-cell lysis and solubilization
Depending on the nature of studied cells and the molecular content that is intended for analyses,appropriate solubilization procedures in small volumes that are approaching the volume of a single cell have to be selected. The extreme variability of biomolecules, possessing different physicochemical properties, renders complete solubilization of all components in one step. Furthermore, processing a large number of cells requires a suitable selection of the specific multi-well micro platforms. It is important to consider common problems of handling small volumes of samples that have to be subsequently manipulated without selective losses of molecules during solubilization and the following separation.
Separation of biopolymers, organic and inorganic components solubilized from single cells
Structural features of biomolecules determine their physicochemical characteristics, such as mass,sequence, charge, hydrophobicity/hydrophilicity,ligand affinity, and conformation. Based on the differences in molecular properties, centrifugation,chromatography, electrophoresis, and solubility methods can be chosen for a variety of combined separations.
Detection, identification, and quantification of all biopolymers, organic and inorganic components from single cells with the single molecular sensitivity
Detection, identification, and quantification of biomolecules present in a sample are based on measurements of signals originating from their interactions with a probe that can be photon, electron,atom, and organic or inorganic molecule. Technological design of analytic instruments that can obtain highquality signal-to-noise ratio from every single molecule in a sample is the prerequisite for quantitative omic measurements. Reaching sensitivity of single molecular detection, identification and quantification can be principally achieved for some classes of biomolecules in individual cells. Technical solutions remain to be the major challenge for single molecular sensitivity in high throughput analyses dealing with hundreds of thousands of cells. In the following section, a more detailed description is reviewed.
Single molecular detection sensitivity in single-cell omics
Single molecules can be detected and identified,either directly or indirectly, using different types of probes. Since the signal output is dependent on both the nature of the probe and the structure of the analyte molecule, identification of the specific type of molecule can be achieved. Various instrumentations,such as spectrometers (mass, optical, ion, magnet),and microscopes (scanning probe, electron, optical,ion), are used for molecular detection and/or identification. The most common types are scanning probes, electron, X-ray, optical (critical angle and super-resolution) microscopes, as well as various spectroscopes measuring mass and/or charge, electrochemical, diffraction, resonance, nuclear magnetic,and paramagnetic properties of molecules. Some of these instrumentations can directly or indirectly detect and identify some types of single molecules using spectral properties of the output signal, and/orviadirect chemical and enzymatic sequencing. If an instrument possesses scanning properties with sufficient lateral and/or depth resolution, the output signal can also be transformed into 2D and 3D images. The limit of instrument detection has to be determined by measurements of signal-to-noise ratio together with proper calibration using standards for each class of molecules. Unfortunately, in high throughput omics analyses, none of the described instrumentation technologies can provide the single molecular detection sensitivity for proteins, glycans,lipids, metabolites, and organic and inorganic compounds in each single cell in tissue, organ, or organism samples until now (Fig. 1B). The exceptions are nucleic acids which can be enzymatically amplified and consequently detected even if there is only a single molecular copy in a single cell (Fig. 1A).
Detection, quantification, and identification of different classes of biopolymer significantly depend on their nature, the number of copies for each type of molecule present in a cell, and molar mass (size) (Fig. 1).In order to simplify the single-cell analyses, and achieve better quantification and identification of biomolecules, the common approach nowadays is partial omics entailing measurements of the content of each class of biomolecules separately.
In single-cell analyses the total number of biomolecules per cell is also referred to either in pg per cell, and/or molar concentration, using the amount in pg and molar mass normalized per single cell volume, and/or moles per cell, as well as part per million, part per billion. The same units are used for expressing a degree of sensitivity and the limit of detection for the particular method and instrument(Fig. 1).
In order to get familiar with a rough range of biomolecule amount to be expected in single cells, and the possibility to detect all of them, approximated biomolecular content range of an average "imaginary"mammalian cell is presented inFig. 1Aand summarized below. The "imaginary" average size human cell would have a volume of 2 pL (20 μm×10 μm×10 μm =2×103μm3)[12], and 70% of water contributing to wet weight. Estimation of the number and concentration for each class of biomolecules is based on experimental measurements, mainly done on cell populations, and on theoretical calculations[13–16]. The "imaginary" single cell would have: 1) 6 to 12 pg of DNA,corresponding to 23 pairs of chromosomes in human,2) 10 to 50 pg of total RNA, and about 0.1 to 0.5 pg of mRNA, corresponding to about 6×105to 3×106molecules with an average mRNA size for coding 40 kDa protein, 3) 20 to 700 pg of proteins, corresponding to about 3×108to 3×109molecules with an average size of 40 kDa, 4) 10 to 50 pg of glycans,corresponding to about 1.2×109to 6×109molecules with an average size of 10 kDa, 5) 10 to 50 pg of lipids, corresponding to about 1.2×109to 6×109molecules with an average size of 5 kDa, and 6) 10 to 20 pg metabolites, corresponding to about 4×109to 8×109metabolites with an average size of 1.5 kDa.
If a cell of 1 pL (1×103μm3) volume contains 1 molecule, which is equivalent to 1.66 ymole, the corresponding concentration will be 1.66 pmol/L. One molecule of the protein with a molar mass of 40 kDa has a weight of 66.4 zg. Typical sensitivity of the current detection level without labeling by various types of mass spectrometry instrumentation for biopolymers and their fragments is about 1 to 10 amole or 40 to 400 fg for the average size 40 kDa protein.This corresponds to about 6×105to 6×106molecules,and for the smaller metabolites of about 1 kDa, it corresponds to a few thousand molecules[17–22]. Any molecule present in a lower copy number would remain undetected (Fig. 1B).
It is very important to note that DNA and mRNA single molecules can be copied by repeating cycles of enzymatic amplification into billions of molecules(Fig. 1A), allowing determination of the original copy number even to the presence of a single molecule in a single cell, as well as their identification by complete sequencing[15,23–24]. For proteins, glycan, and lipid classes of biopolymers, and for metabolites, increasing the number of molecular copies by enzymatic treatments cannot be done until now.
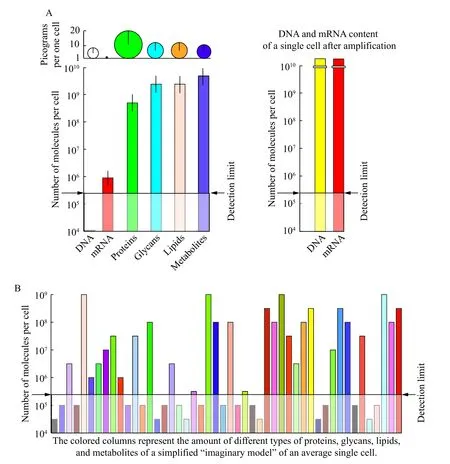
Fig. 1 Biomolecular content of an average "imaginary" mammalian cell with a detection limit for mass spectrometry. A: Copy number of biomolecules and their amount in pg present in a single "imaginary" mammalian cell without and with amplification for nucleic acids. B: Arbitrary graph construction of copy numbers for different types of biopolymers and metabolites, presented as differently colored columns, in an "imaginary" average cell. With the common detection limit of 6×105 molecules for mass spectrometry used in omics analyses,molecules present in lower copy number remain nonexistent.
Large variation of cell sizes ranging from 30 μm3for a sperm cell to 4×106μm3for oocyte[12]imposes another molecular detection and quantification issue at the level of single-cell analyses. Namely, even if a small cell has the same concentration of biomolecules,it will contain a proportionally smaller number of molecules, and thus, it would be more difficult to quantify their ome (Fig. 1). Also, it should be taken into consideration that higher molecular heterogeneity of a single cell leads to more demanding detection and identification of biomolecules. Finally, a very important matter is that every single cell can be analyzed only ones without the possibility for measuring the reproducibility of results with the same cell.Implementation of a variety of labeled internal standards, with a wide range of concentrations, is essential for interpreting small differences between analyzed cells in their molecular content, especially of low abundant biomolecules.
Covalent linking of multiple fluorescence dyes,nanoparticles, and element tags per one molecule can significantly increase sensitivity to the range between 103to 105molecules by optical and/or mass spectroscopy analytical instrumentation, and in some cases even reach the level of single molecule detection for the particulate type of biomolecule[5–11,25–26]. Secondary ion mass spectrometry, as well as direct electrochemical and enhanced optical refractometry with metals can reach levels of single molecular detection sensitivity without labeling, but only in specific cases[27–32]. However, the majority of these methods need further development in order to be routinely applied in single-cell analyses. The above calculations referring to the total number of copies of molecules present in a single cell, and measurements of detection sensitivity based on molecular standard calibration of instruments, confirm that very low abundant species of biomolecules, which are close to the level approaching just one copy per cell, cannot be so far reached in the high throughput analyses of a single cell sample volume (Fig. 1)[17–19].
Overview of single-cell analyses
Cellular heterogeneity
The cellular heterogeneity is intrinsically determined by genetic, epigenetic, and stochastic factors during:a) evolution of unicellular and multicellular organisms,b) embryonal development, and c) adult life cycle.Numerous omics analyses on populations of cells obtained from different tissues, together with classical histological studies, have shown that their average bimolecular contents are different. However, the nature and the extent of variations within such tissuespecific populations of cells is a challenging analytical problem at both cellular and molecular levels. An important scientific problem addressed by single-cell omics is how to obtain complete quantitative information about cellular heterogeneity. The commonly-used classical and most current techniques only permit the incomplete analysis of semi and highly abundant biomolecules usually extracted from more than 1000 cells. Therefore, detection of minor components present in rare occurring cells within a particular population may remain invisible in many analyses, unless each individual cell is processed with the detection sensitivity of a single molecule. Such minor components could belong to the class of important regulatory molecules present during short time periods. Unfortunately, due to technological difficulties, most omics experiments are still done on groups of cells, and some of them may be in different cell cycle stages, and/or physiological states.Collective results, derived from such usually partial omics analyses (genomic, transcriptomic, proteomic,glycomic, lipidomic, and metabolomic), correspond to those of the average cell. Despite these data are not providing information on the heterogeneity of the examined population of cells, they are useful starting point in biomedical research for currently progressing single-cell omics analyses of cell diversity.
Cellomes
The objectives of cellomic analysis are to quantify the total number of cells together with the number of different cell types in an individual and to determine their xenogeneic and allogenic diversity in: a) embryogenesis, b) adult life, and c) pathological cases. Here,two extreme cellome examples, human and wormCaenorhabditis elegans, are summarized.
According to the current extrapolated calculations,an average-sized healthy adult human body has about 3.72×1013cells. These number is derived from the numerous histological analyses of cell sizes, volumes,and their packaging density in tissues and organs[3–4].The extension of this study, based on the multitude of multidisciplinary micro and nano approaches integrating cell and molecular biology, biochemistry,immunology, and microscopy data, leads to the identification of about 400 different cell types in healthy human subjects[33]. These impressive results are shaping human cellome knowledge. To reach the goal of completing the human cellome, measurements of the total molecular content for each individual cell are required. This omic analysis shall integrate all specific biological molecule classes obtained from genomics, transcriptomics, proteomics,glycomics, lipidomics, and metabolomics studies. The current path of developing novel micro and nano methodologies and instrumentation, together with the assembling curated and more complete experimental data obtained from individual tissues and organs derived from all sub-omic disciplines of human cellomics, remains to be very challenging.
The research on the development and cell linage in the male wormC. elegansidentified 1033 cells[34–36].The experimental advantage of this organism, one of few extensively explored model system, has a relatively small size and a low number of cells. These features enable the assembling of the complete cellomes of adult males and hermaphrodites, of all of their embryonal stages, together with all cell linages.All individual cells are identified and anatomically positioned, creating remarkable cell maps of these relatively simple organisms. Larger and more complex multicellular organisms containing billions and/or trillions of cells entail a higher degree of complexity,which inevitably hinders the assembling of their complete cellomes.
Single cell genomics
DNA is an extremely large polymer with the presence of only a few molecules per cell. A human diploid single cell has 2-meter-long DNA strands composed of 3 Gbp[23]. In genomic analyses, DNA identification essentially requires complete sequencing of such large molecules. Current technologies use amplification of DNA molecules present in a single human cell, about 6 to 12 pg per cell, in order to detect nucleotides and DNA fragments during sequencing[13–15,24]. Such enzymatic driven procedure requires complete and errorless copying for hundred percent genome sequencing. Detecting and assessing DNA sequence variations in each individual cell permit assemblies of complete single cell genomes.This information is essential for decoding the functional importance of genomic heterogeneity in somatic and germ cells, as well as those cells rapidly dividing in embryos, in regenerating tissues, in blood cells hematopoiesis, and in tumors.
Results obtained from analyses of single tumor cells demonstrated the evolution of genomic cellular diversity that is associated with tumor growth progression by detection of a variety of single nucleotide mutations[37], exons, and methylomes[38]. Investigation of single cell genome in acute lymphoblastic leukemia reported heterogeneity of clonal cell origins[39]and in triple-negative breast cancer a clonal stasis[40]. Another detailed examination of single nucleotide mutations in 25 tumor cells showed no subpopulation[41]. An interesting publication on human multiple myeloma,isolated from over 200 patients' blood detected:a) changes in copy numbers, b) deletions, and c) high level of mutations[42]. A related study on a single patient tumor biopsy revealed clonal evolution in 58 cells[43]. The dynamics of genome variation were also examined by xenografting of human single cell clones obtained from breast cancer[44]. In neural cells,variation of chromosomal and respective DNA number of copies in mosaic-like fashion was discovered,which specified clonally related differences[45–47].
Single cell transcriptomics
Cell separation, amplification, and sequencing procedures used for genomics are also applied in transcriptomics. They result in the assembly of the complete single cell transcriptomes that are approaching single molecular detection and identification[15].In order to obtain the complete transcriptome for each individual cell, the equal and high level of amplification of all present mRNA molecules is imperative.In spite of being difficult, due to minute amounts of mRNA (0.1 to 0.5 pg) present in a single cell[15], and also due to the rapid mRNA turnover, this single-cell omic field is the most proliferative with over 500 publications according to the curated data[48], Human Cell Atlas[49], and data base from "single cell RNA sequence Data Base[50]that are linked with Gene Expression Omnibus[51]. The number of individual cells analyzed varies from 10 to 1×106in different studies.
Recently constructed "The Single Cell Type Atlas"assembles single cell transcriptomics data collected from 192 individual cell types representing 12 different cell type groups from 13 different human tissues, and correlates them with immunohistochemical analyses of protein expression[52]. Since the end products of mRNA are proteins, transcriptomics data allow the prediction of protein pattern expression that aids in proteomic analyses.
Single cell proteomics
Single cell proteomics is tightly connected with transcriptomics and glycomics. Principally two approaches based on micro- and nano-technologies are used: a) separation of proteins obtained from individual cells either by capillary or 2D gel electrophoresis,and/or analytical chromatography followed by peptide mapping and various types of mass spectrometric identification and quantification[53–58], and b) classical array technology using antibody-tagged probes to detect and identify specific proteins[59–62]. Collected proteomic data are used to generate proteomaps of different species[63].
Since the detection limit for protein with mass spectrometry is about 6×105molecules, and amplification of protein copies cannot be done as nucleic acids, proteomics has presently provided only partial proteomes of single cells. Although impressive progress showing cellular heterogeneity has been achieved, the reported numbers single cells analyzed is at best only a few percent of the total cells present in a tissue and/or organ. Also, thousands of detected and/or quantified and/or identified proteins in single cell proteomics form only a small proportion of the 28 000 different proteins that have been identified in humans. Abundant housekeeping proteins are found very similar in all cell types. Several abundant regulatory proteins show variability among different single cells isolated from various tissues[64–67]. Many less abundant proteins have not been detected and still have unknown variations.
Single cell glycomics
At present, the glycomic field[68]is fast developing but it is still analyzing the population of cells and thus providing us with information about the glycan content of an average cell. Unfortunately, single cell glycomics has not yet been experimentally established.Since glycans are secondary gene products generated by glycosyltransferases catalysis, and are covalently linked either to proteins or lipids, their analyses require specialized approaches different from other biopolymer omics[69–71]. Quantitative single molecular detection and identification of glycans in a single cell remains challenging, because of the enormous variability of over 70 000 glycan structures so far sequenced, and relatively low amounts in the plasma membrane[70–75].
Single cell lipidomics
Lipids are essential biopolymer components of all cellular membranes. As in the case of glycans, their structure is not directly coded in the genome, but it is generated by the catalytic action of series of enzymes.Plasma membrane lipids are often glycosylated.Therefore, lipidomic analyses are tightly connected and overlapping with glycomics studies.
Detection of lipids extracted from the individual cells is performed using different types of mass spectrometric measurements either after chromatographic separation or directly by matrix-assisted time of flight mass spectrometry[10,76], or time of flight mass secondary ion mass spectrometry with the capability of 3D scanning[9,11]. Improvements in sensitivity for analyzing single cells of microorganisms were recently reported[77]. Significant advancement of spatial resolution to the subcellular level lipid analyses of live cells by Raman spectroscopy was also described[78].As discussed above, mass spectrometry has a good level of sensitivity, however, it greatly depends on the type of technology used in the particular instrumentation type, and on the nature of biomolecules present in cellular sample extracts. In praxis, single molecule detection sensitivity for all types of single lipid molecules in single cell extracts has not yet been achieved. However, single cell analytical as well as imaging lipidomics provided very useful comparative studies showing significant cellular heterogeneity in the neural cells[79].
Single cell metabolomics
Metabolites are a structurally highly diverse class of small biomolecules with a molecular mass below 1.5 kDa. Performing single cell metabolomic measurements with single molecular detection is as challenging as those for proteins, glycans, and lipids,since amplification of the signal originating from small molecules is more difficult to achieve than for larger biopolymers. Unfractionated extracts of metabolites from single cells are rapidly identified and quantified either by analytical mass spectrometry or by time of flight secondary ion and/or matrix-assisted mass spectrometry imaging instrumentation with micrometer lateral resolution[80–81]. In order to facilitate the identification of metabolites in complex mixtures, the application of a variety of analytical column chromatography and/or capillary electrophoresis separations directly connected to mass spectrometric analyses is more commonly used. Usually,femtomoles can be detected for a broad range of metabolites, and in some cases for neurotransmitters at several hundred attomoles (about 107molecules)[82].The highest detection level by mass spectrometric application for metabolites is 450 fmol/L by measuring amino acid content and other 40 detectable metabolites within individual single cells obtained from tissue culture[83]. Whether levels of metabolites exist below the detection level, and whether such an amount is relevant for the physiology of cells remains to be answered. This can only be achieved by increasing the detection limit to the single molecular sensitivity for all metabolite species present in the individual mammalian cell with an average size of 2 pL.
Measured levels of cancer-specific metabolites and lipids, administrated anti-cancer drugs, and general metabolites before and after therapeutic treatment indeed showed cellular heterogeneity among individual cancer cells[25,84]. These metabolomics-derived results are useful for choosing and monitoring therapeutic treatments and drug resistance according to fingerprint patterns for the specific metabolites[85–86].
Perspectives of single-cell analyses with single molecular detection: toward the complete omes
Perspective developments in single-cell omics entail technological improvements of: a) micro and nano array devices for high throughput analyses, and b) single molecule detection instrumentation. In 2014,one possible solution for advancement in single-cell omics was proposed and a prototype was constructed using Orsay physics focus ion beam and gas injection system instruments that provide nanometer precision lateral and depths engraving and deposition resolution[27]. This study described the prospect to separate, identify and quantify all molecules present in each of 100 and up to 2500 cells on a single chip of 1 cm in diameter with the single molecular detection sensitivity. Two types of nano in micro prototype devices have been reported[27].
The first device is based on isotachophoresis separation using the novel array design comprising of 100 nano electrophoretic open guides to accommodate parallel processing of 100 different cells on a single 1 cm diameter chip (Fig. 2). This electrophoretic device can separate all types of biomolecules in a single run. The identification is achieved by recording the specific position of biomolecules in a guide upon isotachophoresis. The advantage of the described open guides is founded on their: a) high throughput power,b) capacity to complete separation within one minute,and c) ability to overcome uncontrollable and unpredictable losses of minor molecular compounds adoption associated with capillary electrophoretic and chromatographic devices.

Fig. 2 Nanoelectrophoretic array device. A: Scanning electron micrograph of the complete single noelectrophoretic unit constructed by focus ion beam engraving and gas injection deposition of platinum. B: Scheme of a single nanoelectrophoretic unit (left) and scheme of 10×10 array of nanoelectrophoretic devices (right). C: Engraved micro wells and deposited platinum electrodes. D: Engraved nano channels with different depths, edge softness, and diameters. The figure is adapted in part from ref.[27].
The second device does not involve molecular separation technology but performs parallel analyses of 2500 individual cells by placing each cell in one of 2500 micro wells array per one chip with 1 cm diameter. Each microwell has 250 000 nano wells array with covalently attached specific probes. These probes could be antibodies, aptamers, receptors, and ligands that are either nucleic acids, proteins, glycans,lipids, or organic compounds (Fig. 3). Such nano in micro array theoretically allows omic analyses of all molecules that have specific binding probes, as well as elaborate and quantitative testing for new biomarkers and their corresponding probes.
Besides the novel design of two types of nano in micro devices, single molecular detection sensitivity for each class of biomolecules has also been proposed and shown to be possibly by secondary ion mass spectrometry (SIMS) (Fig. 4)[27]. Using the high energy source of primary ions allows SIMS scanning of nano arrays with a lateral resolution of 50 nanometers[87–88]. During the scanning process of nano in micro array chips, primary ions split analyte biomolecules on the chip surface into their elementary atom that are ionized, then separated and finally detected as secondary ions in a mass spectrometer.The increase in sensitivity is therefore proportional to the number of atoms present in a single molecule. For example, one biopolymer theoretically generates a single signal in a mass spectrometer in classical analyses, using SIMS generation of secondary ions that are atom elements of the same biopolymer would increase the output signal that is proportional to the number of atoms present in that biomolecule.Theoretically, this number would be 24 times higher for a simple monosaccharide containing 24 atoms, and for a smaller glycan containing 5 monosaccharides, it would be 120 times higher. Thus, the increase of biopolymer size leads to proportionally greater sensitivity. Indeed, using serial dilutions of bovine serum albumin (BSA) in polyvinyl alcohol, SIMS is able to quantitatively detect and identify a single BSA protein molecule without labeling (Fig. 4)[27]. BSA has an elemental composition of C2932H4614N780O898S39.Therefore, the spectrum and sum of secondary ion counts originating from a single BSA molecule, even if 10% would be reaching and initiating signal in a detector, permits quantification and identification of a single biopolymer.
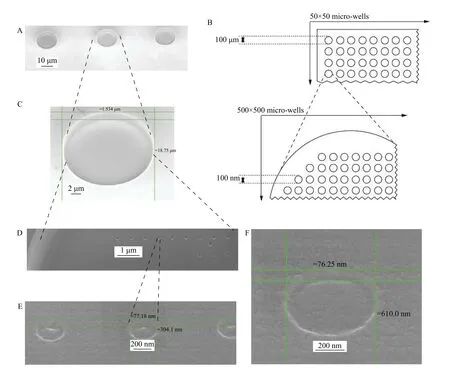
Fig. 3 Schematic drawing and scanning electron microscope images of constructed nano in micro array. A: Three micro wells engraved with a focus ion beam. B: Schematic presentation of nano in micro array device. Scheme of 50×50 array of 100 μm diameter micro-wells (top) and enlarged scheme of a single micro well with an array of 100 nm diameter nano wells inside (bottom). C: Single engraved micro well. D: Engraved nano array within a single micro well. E: Three 300 nm engraved nano wells within a micro well.F: Engraved single 300 nm nano well within a micro well. Horizontal lines indicate approximate depth. Vertical lines indicate diameter in tilted mode scanning electron microscope. The figure is adapted in part from ref.[27].

Fig. 4 Sulfur secondary ion mass spectrometry images of bovine serum albumin serial dilutions. Images represent a single scan with a resolution of 256×256 pixels, where a pixel corresponds to 31.25 nm length (length in nm/number of pixel in this length; 8000 nm/256=31.25 nm. A: 1 mg/mL bovine serum albumin (BSA). B: 0.1 mg/mL BSA. C: 0.02 mg/mL BSA. D: 0.004 mg/mL BSA. Blue dots are clusters of 10 to 1 molecule of BSA. E: 1.6×10−4 mg/mL BSA. Small faint blue dots represent 8 secondary ions counts of sulfur atoms in mass spectrometer detector from a totally of 39 sulfur atoms present in a single BSA molecule. Dot sizes of single and clustered BSA molecules appear larger due to SIMS lateral resolution of about 50 nm. The figure is adapted in part from ref.[27].
Both types of proposed nano in micro devices allow the use of internal and external standards with or without labeling for quantitative single-cell omic analyses with single molecule detection sensitivity.Although proof of concept has been delivered, these devices, methods, and instrumentation still need further development, thoroughly testing, and scalable production of affordable larger quantities.
Acknowledgments
To GNM private funds.
杂志排行

THE JOURNAL OF BIOMEDICAL RESEARCH的其它文章
- Editorial commentary on the special issue of Advances in Nanomedicine
- Superior in vitro anticancer effect of biomimetic paclitaxel and triptolide co-delivery system in gastric cancer
- Nanobody-based immunosensing methods for safeguarding public health
- Bacterial nanocellulose production and biomedical applications
- Hybrid lipopolymer vesicle drug delivery and release systems
- EPR spectroscopy of whole blood and blood components: can we diagnose abnormalities?