Chromatin accessibility profiling provides insights into larval cuticle color and adult longevity in butterflies
2021-10-18Wen-TingWan,Zhi-WeiDong,Yan-DongRen等
Butterflies are diverse in virtually all aspects of their ontogeny,including morphology, life history, and behavior. However, the developmental regulatory mechanisms underlying the important phenotypic traits of butterflies at different developmental stages remain unknown. Here, we investigated the developmental regulatory profiles of butterflies based on transposase accessible chromatin sequencing (ATAC-seq) at three developmental stages in two representative species(Papilio xuthusandKallima inachus). Results indicated that 15%-47% of open chromatin peaks appeared in associated genes located 3 kb upstream (i.e., promoter region) of their transcription start site (TSS). Comparative analysis of the different developmental stages indicated that chromatin accessibility is a dynamic process and associated genes with differentially accessible (DA) peaks show functions corresponding to their phenotypic traits. Interestingly, the black color pattern inP. xuthus4th instar larvae may be attributed to promoter peak-related genes involved in the melanogenesis pathway. Furthermore, many longevity genes in 5th instar larvae and pupae showed open peaks 3 kb upstream of their TSS, which may contribute to the overwintering diapause observed inK. inachusadults.Combined with RNA-seq analysis, our data demonstrated that several genes enriched in the melanogenesis and longevity pathways also exhibit higher expression, confirming that the expression of genes may be closely related to their phenotypic traits. This study offers new insights into larval cuticle color and adult longevity in butterflies and provides a resource for investigating the developmental regulatory mechanisms underlying butterfly ontogeny.
Butterflies are diverse in virtually all aspects of their biology,ranging from morphology, life history, behavior, and biogeography to cellular biology and biochemistry (Baxter et al., 2008; Boggs et al., 2003; Jiggins et al., 2001; Joron &Mallet, 1998; Kunte, 2009; Nijhout, 1991). The study of butterflies has been an integral part of ecology and evolution ever since Darwin and Wallace proposed the theory of natural selection (Poulton, 1890; Shirataki et al., 2010; Wallace, 1865,1871). Wing pattern in adult butterflies is considered as an ideal model system to study evolutionary developmental biology (evo-devo) (Beldade & Brakefield, 2002; John et al.,2011; McMillan et al., 2002; Monteiro, 2015; Reed et al., 2011;Zhang et al., 2017). Nevertheless, few studies have dealt with the molecular mechanism underlying morphological evolution in the developmental stages, e.g., larval and pupal stages,which are important in determining the physiological and morphological traits of adults (Futahashi et al., 2012; Shirataki et al., 2010). For example, in 5th instar larvae, many prepattern genes that guide morphological development in adults are already pre-activated (Reed & Serfas, 2004), and at the pupal stage, the body is completely remodeled for the developing adult (Martin, 2011). Butterflies have evolved various traits in their developmental stages, from outer morphology (such as body color) and internal physiology to cellular and molecular processes, to adapt to their different habitats (Futahashi & Fujiwara, 2006; Futahashi et al., 2012).However, the developmental regulatory mechanisms underlying the many important phenotypic traits of butterflies during their developmental stages remain unknown.
The genetic and molecular basis of phenotypic evolution is a long-standing puzzle in butterfly evolutionary studies. Since Britten & Davidson (1969) proposed the evolutionary model of regulatory sequences, increasing evidence suggests that the evolution of gene regulation leads to the formation of morphological diversity (Carroll, 2000, 2005, 2008; Carroll et al., 2005; Levine & Tjian, 2003). The profiling of open chromatin regions is an effective way to dissect regulatory genomic regions, as chromatin accessibility is relevant to gene expression regulation via the binding of transcription initiation and regulatory elements, especially in promoter regions(Degner et al., 2012; John et al., 2011; Tsompana & Buck,2014). The assay of ATAC-seq was recently developed to assess genome-wide chromatin accessibility using hyperactive Tn5 transposase combined with high-throughput sequencing technology (Buenrostro et al., 2013). It has been applied in several butterfly species to investigate the regulatory evolution of wings and adult brain development(Lewis et al., 2019; Lewis & Reed, 2019; Lugena et al., 2019;Van Der Burg et al., 2019). Based on chromatin accessibility analysis, Lewis et al. (2019) identified fivecis-regulatory elements of theoptixgene underlying color pattern variation inHeliconius. In addition, Van Der Burg et al. (2019) observed dynamic chromatin accessibility duringJunonia coeniawing metamorphosis and found that thespinelessandEcRtranscription factors are associated with changes in accessibility.
To explore the genetic regulatory basis of morphological divergence among butterfly species during development, we investigated chromatin accessibility in three developmental stages (i.e., 4th and 5th instar larvae and pupae) (Figure 1A)in two representative species from two different families(Papilionidae:Papilio xuthus; Nymphalidae:Kallima inachus)using ATAC-seq and RNA-seq. The two selected species,which are distributed in Asia and have high-quality reference genomes available (Li et al., 2015; Nishikawa et al., 2015;Yang et al., 2020), exhibit many morphological and biological differences in the larval, pupal, and adult stages.Papilio xuthuslarvae feed on Rutaceae plants (Zanthoxylum piperitum) and have similar body color and background (black and brown) from the 1st to 4th instar larval stages, but show marked changes at the 5th instar stage, with the development of a green cuticle and reduction of black coloration. This species also exhibits diapause at the pupal stage.Kallima inachuslarvae feed on Acanthaceae plants (Strobilanthes),and usually have a similar body color at all larval stages (1st to 5th instar). This species usually produces three generations each year, with a 5-7 month diapause stage during adulthood showing excellent concealment (Zhou et al., 2005).
Here, the collected samples were first dissected, after which the ATAC-seq and RNA-seq libraries were constructed (see Supplementary Methods). We then obtained high-quality ATAC-seq data for 4th instar larvae (L4), 5th instar larvae(L5), and pupae (P) for the two butterfly speciesP. xuthus(Px)andK. inachus(Kin) (Supplementary Table S1). Based on ATAC-seq data analysis, dynamic chromatin accessibility peaks appeared during the different developmental stages in both species (peak numbers, Px: 20 367 (L4), 9 648 (L5), 12 001(P); Kin: 28 751 (L4), 85 433 (L5), 105 067 (P)) (Table 1). We then performed comparative assays between stages for each species, i.e., L4 vs. L5, L4 vs. P, and L5 vs. P (Supplementary Table S2) to identify changes in DA peaks. Results indicated that some accessible sites became stronger or weaker between stages, i.e., showing increased or decreased DA peaks, respectively. Changes in DA peaks were identified for each instar pair in bothP. xuthusandK. inachus(Px: 1 212-5 208; Kin: 24 755-30 979) (fold-change≥2,P≤0.05)(Supplementary Table S2), suggesting dynamically changing chromatin accessibility in the different developmental stages.By analyzing the distribution of peaks in the corresponding reference genomes (Nishikawa et al., 2015; Yang et al.,2020), 15%-47% of chromatin accessibility peaks and 11%-36% of DA peaks appeared in associated genes located 3 kb upstream of their TSS regions (Supplementary Tables S3, S4), and thus can be considered as promoter peaks(Foissac et al., 2019), which play important roles in the transcription of associated genes. These promoter peaks showed consistent signals at the TSS regions in all developmental stages in both species (Supplementary Figure S1). We also observed some ATAC-seq peaks in gene bodies(either introns or exons) and distal intergenic regions(Supplementary Tables S3, S4). These results indicate that ATAC-seq technology can efficiently detect both proximal and distal intergenic regulatory regions in butterflies.
The observed peaks are open chromatin regions, which may contain transcription factor binding sites (TFBSs) (Van Der Burg et al., 2019). TFBSs play important roles in the regulation of gene expression mediated by transcription factors (TFs). Therefore, we identified the representative motifs of TFBSs in accessible peaks and predicted their potential TFs in each developmental stage and instar pair using MEME software (v5.3.2) (Machanick & Bailey, 2011).We identified 14, 11, and eight motifs in 4th instar larvae, 5th instar larvae, and pupae ofP. xuthus, respectively, and 14, 11,and nine motifs in 4th instar larvae, 5th instar larvae, and pupae ofK. inachus, respectively (Table 1; Supplementary Table S5). More motifs in DA peaks were identified inK.inachus(12 motifs in L4 vs. L5 and 14 motifs in L4 vs. P and L5 vs. P) than inP. xuthus(nine motifs in L4 vs. L5, 10 motifs in L4 vs. P, and eight motifs in L5 vs. P) (Supplementary Tables S2, S6). After comparing the identified motifs with known motifs in the JASPAR database (http://jaspar.genereg.net/) using TomTom software (v5.3.2) (Machanick &Bailey, 2011), we mined the potential TFs after combining all motifs. In total, 71 TFs were predicted in the three developmental stages (Supplementary Figure S2; Table 1;Supplementary Tables S5, S7) and 43 TFs binding to TFBSs were predicted within the DA ATAC-seq peaks among the instar pairs in both butterfly species (Supplementary Tables S2, S6, S8). We further analyzed the position of the DA peaks including TFBSs related to these TFs and found that many TFs bind to the promoter regions of genes (Supplementary Table S9). The TFs binding to the DA peaks may therefore regulate morphological changes between stages.

Table 1 Summary of ATAC-seq peaks and peak-associated genes and motifs of transcription factor binding sites (TFBSs) in open peaks and transcription factors (TFs) at three developmental stages in Papilio xuthus (Px) and Kallima inachus (Kin)
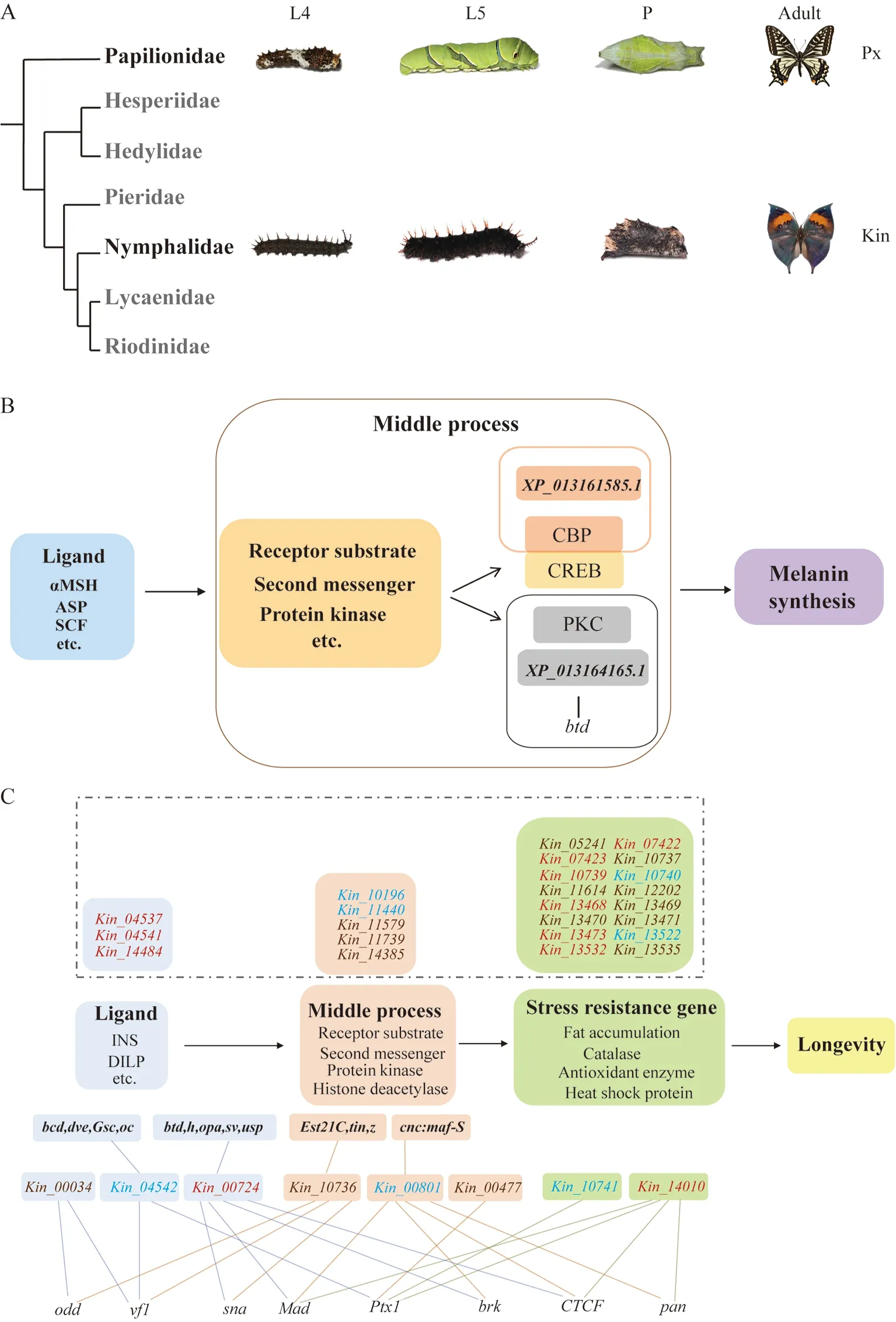
Figure 1 Species studied and schematic of KEGG pathways showing position of associated genes to differentially accessible (DA)ATAC-seq promoter peaks and predicted transcription factors (TFs) at their promoter regions in Papilio xuthus (Px) and Kallima inachus(Kin)
Our ATAC-seq data provide important insights into the molecular mechanism underlying larval color pattern changes in 4th instar larvae (L4) to 5th instar larvae (L5) inP. xuthus(i.e., from black to green) (Figure 1A). Larval pattern switch is an important adaptive strategy and regulated by juvenile hormones during the early L4 stage (JH-sensitive period)(Futahashi & Fujiwara, 2008). Kyoto Encyclopedia of Genes and Genomes (KEGG) enrichment analysis indicated that the DA ATAC-seq promoter peak-associated genes(XP_013161585.1andXP_013164165.1) in L4 vs. L5 were significantly related in the melanogenesis pathway (ko04916)(Supplementary Figure S3A and Table S10). However, we failed to identify hormone-related genes with the DA ATACseq promoter, which may be because hormones often play key roles during specific developmental time periods, and our sampling focused on different instar larvae, not specific time points. DA ATAC-seq peak analysis showed that the accessible promoter peaks ofXP_013161585.1had strong signals in the L4 stage but no signal in the L5 stage(Supplementary Table S11). The accessible promoter peaks ofXP_013164165.1had signals in both the L4 and L5 stages,but the signals were stronger in L5 (Supplementary Table S11). Based on motif and TF analysis in L4 vs. L5 inP.xuthus, one motif (CCYWYCCCYHYHMCYACCCCC) and one TF (btd) were identified on the promoter region of the geneXP_013164165.1, while no motif or TF was identified on the geneXP_013161585.1(Figure 1B; Supplementary Table S12). Combined with RNA-seq analysis,XP_013161585.1andXP_013164165.1showed slightly higher expression in L4 than in L5 (Supplementary Figure S4A), suggesting that high promoter peak accessibility may promote gene expression.Furthermore,XP_013161585.1is annotated as a histone acetyltransferase (HAT) p300 and binds to Creb-binding protein (CBP), constituting an important regulatory factor in the Wnt signaling pathway (Li et al., 2007). The signaling ligand WntA, a member of the Wnt family, controls variation in melanin patterning acrossHeliconiusand other butterfly species (Kronforst & Papa, 2015; Van Belleghem et al., 2020).XP_013164165.1is annotated as protein kinase C (PKC) and indirectly promotes the synthesis of melanin (Figure 1B). Our data indicate that the two genes and open peaks in their promoter regions likely play important roles in the black patterns of the epidermis in 4th instar larvae ofP. xuthus. We checked the chromatin accessibility patterns for several wellknown genes (ebony,black,laccase,prophenoloxidase, andyellow) encoding enzymes involved in melanin biosynthesis.Results showed that the genes exhibited different open peaks in the three developmental stages inP. xuthus(Supplementary Table S13).
Interestingly, ATAC-seq analysis also provided molecular evidence for the longevity of diapaused adults. Butterflies can hibernate, in the form of diapause, during different developmental stages, but most commonly during the larval and pupal stages (Danks, 1987). Previous study on 290 butterfly species from North America found that diapause is more common in the larval and pupal stages (58% and 24%,respectively) than in the egg and adult stages (13% and 5%,respectively) (Scott, 1979). Further research on 110 Lepidopteran species reported similar results, with diapause predominantly occurring in the larval and pupal stages (93%)and more limitedly in the egg and adult stages (7%)(Saunders, 2002). For example, egg diapause is common inParnassiusspecies, such asP.apollo,P.bremeri, andP.glacialis(Elwes, 1886; Muscarella, 2010; Weiss, 1999); larval diapause is common in certain Nymphalidae species, such asLimenitis Archippus, and in several Pieridae species, such asColias alexandra(Clark & Platt, 1969; Hayes, 1982); pupal diapause is most common in Papilionidae species, such asPapilio xuthus, and in Pieridae species, such asPieris rapae(Kaneko & Katagiri, 2004; Yamanaka et al., 2004); and adult diapause is found in Nymphalidae species, such asK. inachusandPolygonia c-aureum(Hiroyoshi & Reddy, 2018; Protas &Patel, 2008; Zhou et al., 2005). Based on KEGG enrichment analysis, we identified severalK. inachusgenes enriched in the longevity regulating pathway (ko04213) in the L4 vs. L5(18 genes) and L4 vs. P (25 genes) comparisons, which were distributed throughout the whole pathway (Supplementary Figure S3B, C and Table S10). In addition, we identified five motifs and 17 TFs on the promoter regions of five out of the 18 identified genes in L4 vs. L5, and five motifs and seven TFs on the promoter regions of three out of the 25 identified genes in L4 vs. P (Figure 1C; Supplementary Table S12). Among the 18 genes identified from the L4 vs. L5 comparison, two DA promoter peaks of one gene (Kin_11440) showed significantly stronger signals in L4 larvae, but no signal in L5 larvae, while another 18 DA promoter peaks near 17 genes showed significantly stronger signals in L5 (Supplementary Table S11). Based on combined RNA-seq analysis, three genes (Kin_10196,Kin_11440, andKin_13522) out of the 18 identified genes showed significantly higher expression in L5(Supplementary Figure S4B).Kin_13522is annotated as an alpha crystallin/heat shock protein, which comes from a family of large and dynamic oligomers that are highly expressed in long-lived muscle, lens, and brain cells (Augusteyn, 2004;Bagneris et al., 2009). Among the 25 genes identified from the L4 vs. P comparison, two DA promoter peaks near two genes showed significantly strong signals in the L4 stage but no signal in the P stage, while 29 DA promoter peaks in 25 genes showed significantly stronger signals in the P stage(Supplementary Table S11). Based on combined RNA-seq analysis, six (Kin_07423,Kin_13471,Kin_13473,Kin_13532,Kin_13535, andKin_14010) out of the 25 identified genes showed significantly high expression in the P stage(Supplementary Figure S4C). Several of these highly expressed genes (Kin_07423,Kin_13473,Kin_13532,Kin_13535, andKin_13471) are annotated as alpha crystallin/heat shock proteins and heat shock protein 70,which are closely related to organismal growth and development, metabolic activity, apoptosis, and longevity(Bagneris et al., 2009; Lee et al., 2005; Seddigh, 2019;Shilova et al., 2018; Vos et al., 2016). Comparing the larval and pupal stages ofK. inachus, we found that more genes were enriched in the longevity regulating pathway in pupae.These data suggest that, compared with the L4 stage, more accessible promoter peaks with longevity-related genes were open in the L5 and P stages, and most showed higher expression. Notably, the number and expression of genes enriched in the longevity regulating pathway were markedly higher in the pupae ofK. inachus. The results demonstrate that adult diapause and overwintering could be attributed to the regulatory expression of longevity genes in 5th instar larvae and pupae. Thus, our data lay a novel foundation for future investigations on adult overwintering in butterflies.
Taken together, we revealed the chromatin accessibility profiles of three developmental stages in two butterfly species from Papilionidae and Nymphalidae. Our results showed that chromatin accessibility is a dynamic process across all developmental stages and is highly correlated with phenotypic traits. Furthermore, we showed that two genes in 4th instar larvae, which function in the melanogenesis pathway, had open peaks at their promoter regions and may play key roles in the cuticle color of such larvae inP. xuthus. We also found that longevity in diapaused adults may benefit molecularly from the expression of longevity genes in 5th instar larvae and pupae. This study not only provides novel insights into larval cuticle color and adult longevity, but also provides an important data resource for investigating developmental regulatory mechanisms in butterfly ontogeny.
DATA AVAILABILITY
All ATAC-seq and RNA-seq data for the three developmental stages (in fastq format) used in this study have been deposited in the Sequence Read Archive of NCBI with Accession Nos. SRP313417 and SRP313418.
SUPPLEMENTARY DATA
Supplementary data to this article can be found online.
COMPETING INTERESTS
The authors declare that they have no competing interests.
ACKNOWLEDGEMENTS
We would like to thank Prof. Yong Wang for suggestions in designing this work.
AUTHORS’ CONTRIBUTIONS
X.Y.L. and W.W. conceived the study, designed the scientific objectives, led the project, and prepared the manuscript.W.T.W. conducted library construction and data analyses.Y.D.R., J.Y., X.Y.P., W.L., J.W.H., and C.Y.M. helped with data analyses. Z.C., W.T.W., Z.W.D., G.C.L., and R.P.Z.raised the butterflies and prepared RNA samples for genomic sequencing. Z.W.D., J.W.H., P.H., and J.L. helped with manuscript preparation. W.T.W., X.Y.L., and W.W. wrote the manuscript. All authors read and approved the final version of the manuscript.
杂志排行

Zoological Research的其它文章
- Phylogenomic relationships and molecular convergences to subterranean life in rodent family Spalacidae
- Prosecution records reveal pangolin trading networks in China, 2014-2019
- Chromosome-scale genome assembly of brownspotted flathead Platycephalus sp.1 provides insights into demersal adaptation in flathead fish
- Deletion of phosphatidylserine flippase β-subunit Tmem30a in satellite cells leads to delayed skeletal muscle regeneration
- Role of juvenile hormone receptor Methoprene-tolerant 1 in silkworm larval brain development and domestication
- Captopril alleviates lung inflammation in SARS-CoV-2-infected hypertensive mice