Role of juvenile hormone receptor Methoprene-tolerant 1 in silkworm larval brain development and domestication
2021-10-18YongCuiZuLianLiuCenCenLiXiangMinWeiYongJianLinLangYouZiDanZhuHuiMinDengQiLiFengYongPingHuangHuiXiang
Yong Cui, Zu-Lian Liu, Cen-Cen Li, Xiang-Min Wei, Yong-Jian Lin, Lang You, Zi-Dan Zhu, Hui-Min Deng,Qi-Li Feng,*, Yong-Ping Huang,*, Hui Xiang,*
1Guangdong Provincial Key Laboratory of Insect Developmental Biology and Applied Technology, Guangzhou Key Laboratory of Insect Development Regulation and Application Research, Institute of Insect Science and Technology, School of Life Sciences, South China Normal University, Guangzhou, Guangdong 510631, China
2 Key Laboratory of Insect Developmental and Evolutionary Biology, Institute of Plant Physiology and Ecology, Shanghai Institutes for Biological Sciences, Chinese Academy of Sciences, Shanghai 200032, China
3 College of Life Sciences, Xinyang Normal University, Xinyang, Henan 464000, China
ABSTRACT The insect brain is the central part of the neurosecretory system, which controls morphology,physiology, and behavior during the insect’s lifecycle.Lepidoptera are holometabolous insects, and their brains develop during the larval period and metamorphosis into the adult form. As the only fully domesticated insect, the Lepidoptera silkworm Bombyx mori experienced changes in larval brain morphology and certain behaviors during the domestication process. Hormonal regulation in insects is a key factor in multiple processes.However, how juvenile hormone (JH) signals regulate brain development in Lepidoptera species,especially in the larval stage, remains elusive. We recently identified the JH receptor Methoprene tolerant 1 (Met1) as a putative domestication gene.How artificial selection on Met1 impacts brain and behavioral domestication is another important issue addressing Darwin’s theory on domestication. Here,CRISPR/Cas9-mediated knockout of Bombyx Met1 caused developmental retardation in the brain, unlike precocious pupation of the cuticle. At the whole transcriptome level, the ecdysteroid (20-hydroxyecdysone, 20E) signaling and downstream pathways were overactivated in the mutant cuticle but not in the brain. Pathways related to cell proliferation and specialization processes, such as extracellular matrix (ECM)-receptor interaction and tyrosine metabolism pathways, were suppressed in the brain. Molecular evolutionary analysis and in vitro assay identified an amino acid replacement located in a novel motif under positive selection in B. mori,which decreased transcriptional binding activity. The B. mori MET1 protein showed a changed structure and dynamic features, as well as a weakened coexpression gene network, compared with B.mandarina. Based on comparative transcriptomic analyses, we proposed a pathway downstream of JH signaling (i.e., tyrosine metabolism pathway) that likely contributed to silkworm larval brain development and domestication and highlighted the importance of the biogenic amine system in larval evolution during silkworm domestication.
Keywords: Met1; Brain; Artificial selection;Tyrosine metabolism pathway; Silkworm;
INTRODUCTION
The insect brain is the central part of the neurosecretory system, which controls morphology, physiology, and behavior during the insect’s lifecycle. Lepidoptera are holometabolous insects, which experience brain development during the larval period and metamorphosis. Larval brain size increases after each molt and brain morphology changes markedly during metamorphosis, including the differentiation of optic lobes,antennal lobes, and mushroom bodies (Champlin & Truman,1998).
Two key insect hormones, i.e., juvenile hormone (JH) and ecdysteroid (20-hydroxyecdysone, 20E), orchestrate insect growth, molting, metamorphosis, and reproduction via their receptors and responsive genes (Liu et al., 2018). JH prevents 20E-induced larval-pupal/adult metamorphosis until insects reach the appropriate stage and is therefore referred to as the“status quo” hormone (Daimon et al., 2012, 2015; Liu et al.,2018). In many insect species, Methoprene-tolerant (Met), a member of the basic helix-loop-helix-Per-Arnt-Sim transcription factor family, functions as a JH receptor and positively regulates the expression of the JH-responsive geneKrüppel homolog 1(Kr-h1) (Daimon et al., 2015; Li et al.,2019; Zhu et al., 2019). During the larval stages,Kr-h1represses the pupal-specifier geneBroad-complex(Br-C)(Kayukawa et al., 2016) as well as the adult-specifier geneEcdysone-induced protein 93(E93) (Kayukawa et al., 2017),which can be induced by 20E, to prevent precocious larvalpupal or larval-adult transition.
The mechanism of JH regulation varies among tissues and species. For example, recent advances indicate that in the reproductive system, the role of JH cannot be explained simply as “status-quo” in many tissues and the role of JH signaling shows notably variation among species (Li et al.,2019; Riddiford, 2020; Shpigler et al., 2020). In the brain,studies onDrosophilaindicate that depletion of JH or knockout of its receptors can repress optic lobe proliferation and cause premature abnormalities in the organization of optic lobe neuropils during larval-pupal transition (Abdou et al., 2011;Riddiford et al., 2010). In these studies, the precocious appearance of the ecdysone receptor (EcR) or expression of its responsive gene (Br-C) was detected, suggesting that JH is required for the development of the nervus opticus and the prevention of 20E signaling in the brain (Abdou et al., 2011;Riddiford et al., 2010). JH also drives the maturation of spontaneous mushroom body neural activity and behavior in adultDrosophila(Leinwand & Scott, 2021). Similar functions have also been found in hemimetabolous house crickets(Cayre et al., 1994, 2005).
The role of JH/JH signaling in Lepidoptera brain development is still largely unknown compared with the role of ecdysone, which is reported to induce cell proliferation during optic lobe neurogenesis in the mothManduca sexta(Champlin& Truman, 1998). Recent research showed that application of a potent JH analogue in 5th instar silkworm larvae stimulates the division of brain neurosecretory cells, thereby implying that JH plays a role in the functional development of the silkworm brain (Tanriverdi & Yelkovan, 2020). This raises the question of whether (and how) JH signaling regulates brain development in Lepidoptera species. In the current study, we used the domestic silkwormBombyx morias a model and performed CRISPR/Cas9-mediated knockout of the JH receptorMet1. We then conducted comparative transcriptome analyses in the brain and epidermis to decipher possible features of JH signaling in the brain.
Bombyx moriis a fully domesticated insect andMet1has been identified as a candidate domestication gene (Xiang et al., 2018), thus providing a good opportunity the address the functional impact of the JH receptor from an evolutionary view.Similar to the decrease in brain size reported in some domesticated animals (Agnvall et al., 2017; Stuermer &Wetzel, 2006),B.morialso shows a “simpler” larval brain, with weak brain lobe fusion and smaller relative brain volume compared with its wild ancestorB.mandarina(Figure 1). This suggests that artificial selection may impact the brain. In addition, like other domestic animals (Axelsson et al., 2013;Wang et al., 2016), selection on brain and behavior, mediated by the neuronal system (Pennisi, 2011), is reported to have occurred during silkworm domestication (Xiang et al., 2018). In regard to behavior,B.morishows increased hyperphagia,reduced locomotor activity, loss of escape response, and greater tolerance to human handling compared withB.mandarina(Mignon-Grasteau et al., 2005; Nusinovich et al.,2017). Given that JH may influence insect behavior patterns through regulation of brain neurogenesis and neural activity as well as neurotransmitter signaling (Leinwand et al., 2021;Sasaki et al., 2012; Wheeler et al., 2015), this raises theBombyx moriis a fully domesticated insect andMet1has been identified as a candidate domestication gene (Xiang et al., 2018), thus providing a good opportunity the address the functional impact of the JH receptor from an evolutionary view.Similar to the decrease in brain size reported in some domesticated animals (Agnvall et al., 2017; Stuermer &Wetzel, 2006),B.morialso shows a “simpler” larval brain, with weak brain lobe fusion and smaller relative brain volume compared with its wild ancestorB.mandarina(Figure 1). This suggests that artificial selection may impact the brain. In addition, like other domestic animals (Axelsson et al., 2013;Wang et al., 2016), selection on brain and behavior, mediated by the neuronal system (Pennisi, 2011), is reported to have occurred during silkworm domestication (Xiang et al., 2018). In regard to behavior,B.morishows increased hyperphagia,reduced locomotor activity, loss of escape response, and greater tolerance to human handling compared withB.mandarina(Mignon-Grasteau et al., 2005; Nusinovich et al.,2017). Given that JH may influence insect behavior patterns through regulation of brain neurogenesis and neural activity as well as neurotransmitter signaling (Leinwand et al., 2021;Sasaki et al., 2012; Wheeler et al., 2015), this raises the question of whether (and how) artificial selection of the JH receptorMet1influenced changes in the brain and behavior during silkworm domestication. Thus, we conducted evolutionary and functional analyses of amino acid replacement inB.moriand performed comprehensive brain transcriptome analyses betweenB.moriandB.mandarina.
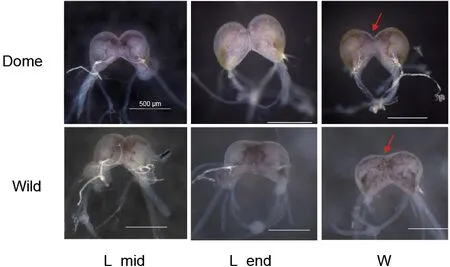
Figure 1 Comparison of brain morphology in B. mori and B.mandarina at larval stage
Our results showed that unlike preventing precocious metamorphosis of the cuticle, JH signals mediated byMet1potentially promoted silkworm larval brain development,without obvious repression of pupal- and adult-specifier genes. Furthermore, two JH downstream signaling pathways,i.e., tyrosine metabolism and extracellular matrix (ECM)-receptor interaction pathways, likely contributed to silkworm larval brain development. Artificial selection ofMet1fixed two nonsynonymous sites specific inB.mori, which affected protein structure and weakened transcriptional binding activity and the co-expression network ofMet1. In addition, the biogenic amine system may be involved in the changes in larval behavior during silkworm domestication.
MATERIALS AND METHODS
Silkworm strains
Domestic (B.mori, strain P50) and wild silkworms (B.mandarina) were used for all experiments and comparative transcriptomic analyses. TheB.mandarinaspecimens were collected from Zhejiang Province (China) and were maintained under laboratory conditions. The multivoltine silkworm strainNistariwas used for CRISPR-Cas9 knockout. Larvae were reared on fresh mulberry leaves under standard conditions(Tan et al., 2013).
CRISPR-Cas9-mediated knockout of Met1
The target sequence was designed according to the feature of GGN19GG (Fu et al., 2014; Hwang et al., 2013). Three 23 bp sgRNA targeting sites were identified at the exon ofMet1and were used for knockout (Supplementary Figure S1). The sgRNA DNA template was synthesized by polymerase chain reaction (PCR), with Q5® High-Fidelity DNA Polymerase (New England Biolabs, USA). One oligonucleotide (Met1-228-sgF1,Met1-563-sgF2, andMet1-899-sgF3), which encoded the T7 polymerase binding site, sgRNA targeting sequence, and overlap sequence, was separately annealed to a common oligonucleotide that encoded the remainder of the sgRNA sequence (sgRNA-R). The reaction conditions were the same as described previously (Cui et al., 2018).
The sgRNA was synthesized based on the DNA templatein vitrowith a MAXIscript® T7 Kit (Ambion, USA) according to the manufacturer’s instructions. The transcribed sgRNA was precipitated by phenol/chloroform/isoamylol (25:24:1, pH<5.0,Solarbio, China) and by isopropanol, then quantified using a NanoDrop-2000, re-dissolved in RNase-free water to 1 000 ng/μL, and finally stored at −80 °C. The Cas9 gene template was provided by the Shanghai Institute of Plant Physiology and Ecology (China). Cas9 mRNA was prepared using a mMESSAGE mMACHINE® T7 kit (Ambion, USA) according to the manufacturer’s instructions. The transcribed sgRNA was precipitated by phenol/chloroform/isoamylol (25:24:1, pH<5.0,Solarbio, China) and quantified using a NanoDrop-2000,diluted to 1 000 ng/μL in RNase-free water, and stored at−80 °C.
TheB.morieggs were collected and injected within 6 h after oviposition. Cas9 mRNA (1 000 ng/μL) and sgRNA(1 000 ng/μL) were mixed and injected into preblastodermNistariembryos (~12 nL/egg) using a micro-injector(FemtoJet®, Germany), according to the standard protocols.The injected eggs were incubated in a humidified chamber at 25 °C for 9-10 days until hatching.
Target site activity examination by in vitro DNA cleavage assay
To test the cleavage activity of selected target sites, a DNA cleavage assay as performed. A mix of 150 ng of Cas9 protein(New England Biolabs, China), 200 ng of sgRNA, 2 μL of 10×NEB buffer, and 100 ng of plasmid containing a target sequence was made and incubated at 37 °C for 1 h. After heating to inactivate Cas9 protein at 65 °C for 10 min, the reaction mixture was analyzed in 1% agarose gel.
Genomic DNA extraction and genotyping analysis
Genomic PCR and sequencing were carried out to examine theMet1mutation induced by the CRISPR/Cas9 system.Genomic DNA was extracted using DNA extraction buffer(2.5:1:2:2.5 ratio of 10% SDS to 5 mol/L NaCl to 0.5 mol/L EDTA to 1 mol Tris-HCl, pH=8) and incubated with 60 mg/mL proteinase K, then purified via standard phenol-chloroform and isopropanol precipitation extraction, followed by RNaseA treatment. The PCR conditions were as follows: 94 °C for 2 min, 35 cycles of 94 °C for 30 s, 57 °C for 30 s, and 72 °C for 50 s, followed by a final extension of 72 °C for 10 min. The PCR products were cloned into a pMDTM19-T Vector (Takara,Japan). The primers designed to detect mutagenesis in the second targeted site are listed in Supplementary Table S1.
RNA extraction, cDNA synthesis, and quantitative realtime PCR (qRT-PCR)
The epidermis and brains of wild-type (WT) and mutant 4thinstar silkworms were dissected. Total RNA was extracted using TRIzol reagent (TaKaRa, China) according to the manufacturer’s instructions. After purification with phenolchloroform, 2 μg of RNA was treated with 2 units of DNase I to remove trace amounts of genomic DNA. Reverse transcription was performed using the Reverse Transcriptase M-MLV Kit according to the manufacturer’s protocols (TaKaRa, China).The primers used for amplifyingBmKr-h1cDNA were 5'-ACCCATACTGGCGAGCGACCAT-3'(forward) and 5'-CCTC TCCTTTGTGTGAATACGACGG-3'(reverse). The primers used for cDNA amplification ofBmRp49(internal control) were 5'-CTCCCTCGAGAAGTCCTACGAACT-3' (forward) and 5'-TGCTGGGCTCTTTCCACGA-3' (reverse). The qRT-PCR was performed under the following conditions: SYBR Premix Ex Taq (2×) (Promega, USA): 10 μL in 20 μL reaction volume;primer concentrations: 0.4 μL (10 μmol/L); immunoprecipitated DNA samples: 4 μL. The mixtures were incubated at 95 °C for 10 s, then 40 cycles at 95°C for 5 s and 60 °C for 31 s using an ABI7300 fluorescence quantitative PCR system (Applied Biosystems, USA). Error bars represent standard deviation(SD) of three replicates. Significant differences were analyzed by Student’st-test.*,**, and***indicate false discovery rate(FDR)-correctedP<0.05, 0.01, and 0.001, respectively.
RNA sequencing (RNA-seq) and data analysis
Three duplicate samples were set for RNA-seq of the WT and mutant epidermis and brain samples and the domestic andB.mandarinabrain samples, respectively. The epidermis and brain samples were dissected, stored in dry ice, and then sent to the Novogene Company (China) for RNA extraction and RNA-seq. Sequencing libraries were generated using a NEBNext® UltraTM RNA Library Prep Kit for Illumina® (New England Biolabs, USA) following the manufacturer’s recommendations and index codes were added to attribute sequences to each sample. Clustering of the index-coded samples was performed on a cBot Cluster Generation System using a HiSeq 4000 PE Cluster Kit (Illumina, USA) according to the manufacturer’s instructions. After cluster generation, the library preparations were sequenced on the Illumina Hiseq 4000 platform (Illumina, USA) and 150 bp paired-end reads were generated.
Sequenced raw data were qualified, filtered, built, and mapped to the reference silkworm genome database (http://www.silkdb.org/silkdb/) using TopHat v2.1.1 (Trapnell et al.,2009). HTSeq v0.6.0 was used to count read numbers mapped to each gene and further normalized using DEseq2 in the R package v1.16.1 (TNLIST, China) (Anders et al., 2015;Love et al., 2014). Principal component analysis (PCA) was performed using genes expressed in the brain (normalized reads>5) with online software (https://www.omicshare.com/tools/).
Differentially expressed genes (DEGs) were identified using Cuffdiff in the package Cufflinks v2.2.1 (Ghosh & Chan, 2016),with correctedP<0.05 and |Log2foldchange|>1. The expression value of each gene was calculated and normalized using fragments per kilobase of exon per million reads mapped(FPKM) (Trapnell et al., 2010). Gene ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway enrichment analyses of DEGs were implemented online(https://www.omicshare.com/tools/) using all expressed genes(FPKM>1) in the brain and cuticle of the silkworm as background.
Identification of selective sweep
Based on available whole-genome single nuclear polymorphic(SNP) data of the domesticated andB.mandarinapopulations(https://doi.org/10.5061/dryad.fn82qp6), the selection signature ofMet1was screened according to the pipeline applied in Xiang et al. (2018) (https://doi.org/10.5061/dryad.fn82qp6). Specifically, a sliding window approach with 5 kb windows sliding in 500 bp steps was applied to identify genomic regions associated with domestication. To discern selective sweeps from the potential background caused by the bottleneck effect, a very stringent threshold was set to screen out regions that significantly deviated from the overall distribution. Only those windows within the top 1% of selective signatures (correspondingP-value ofZtest<0.001) and appliedFst (fixation index) between the two groups were used to represent the selective signatures, taking the highest 1%value as the cutoff. Selection in theB.morigroup (i.e., early domesticated group) was further confirmed by limitingπat a relatively low level (lowest 5%) and reduction of diversity(ROD) (1-πdomestic/πwild; representing reduction of diversity in domesticated lines). Allelic frequency and SNP annotation were calculated using in-house Perl scripts.
Examination of amino acid substitution and remodeling of 3D structure of MET1
The amino acid sequences of MET1 inB.mori(GenBank accession No. NM_001114986.1) were independently aligned with those ofB.mandarina(https://doi.org/10.5061/dryad.fn82qp6),Antheraea yamamai(Kim et al., 2018),Manduca sexta(Kanost et al., 2016),Spodoptera litura(GenBank accession No. XP_022837764.1),Helicoverpa armigera(GenBank accession No. XP_021188262.1),Plodia interpunctella(GenBank accession No. ANZ54967.1),Amyeloistransitella(GenBank accession No.XP_013186860.1),Operophtera brumata(GenBank accession No. KOB74415.1), andDendrolimus spectabilis(GenBank accession No. ATB19377.1) using MEGA 6.0 (Tamura et al.,2013). To explore the structural effects of amino acid residue replacement, the 3D structures of theB.moriandB.mandarinaMET1 proteins were remodeled using Protein Structure Prediction Server (PS)2v3.0 with default parameters(http://ps2v3.life.nctu.edu.tw/) (Huang et al., 2015). The structures were saved in PDB format and analyzed using Swiss-PdbViewer v4.0.1 (Swiss Institute of Bioinformatics,Lausanne, Switzerland) (Guex & Peitsch, 1997). With the predicted 3D structure and query sequences, the packing density of each residue was calculated by the weighted contact number (WCN) derived from protein dynamical properties (Lin et al., 2008).
Phylogenetic analysis by maximum-likelihood (PAML) on Met1
The rate ratio (ω) of nonsynonymous to synonymous nucleotide substitutions was estimated using PAML v4.8(Yang, 1997). After high-quality codon alignment of the above sequences, a series of evolutionary models in the likelihood framework were compared using the species tree of the insects. One ratio model was used to detect average ω across the tree (ω0). Two ratio branch models and two ratio branchsite models were used to detect the ω of the appointed branch to test the (ω1) and ω of all other branches (ω_background).A likelihood ratio test was performed to compare the fit of the two ratio models with the one ratio model to determine whetherMet1is rapidly evolved in the appointed branch(ω1>ω0; ω1>ω_background;P-value<0.05). The branch site model was also used to detect amino acid sites likely to be rapidly evolved in the appointed branch using Bayes Empirical Bayes (BEB) analysis (Yang et al., 2005).
Production of proteins in vitro
A full-length MET1 open reading frame (ORF) fragment was amplified using cDNA of the silk gland from domestic andB.mandarinasilkworms with primers: Met-CDS-SgfI-F and Met-CDS-PmeI-R (Supplementary Figure S1 and Table S1).Obtained DNA fragments were then sub-cloned into the pF25A ICE T7 Flexi vector between the SgfI and PmeI restriction enzyme sites, generating recombinant expression vectors. The nucleic acid sequence corresponding to the region between the two Per-Arnt-Sim (PAS) domains was truncated and sent to TSINGKE (China) for artificial synthesis.The truncated fragment was also sub-cloned into the pF25A ICE T7 Flexi vector, as described above.
Recombinant proteins were produced using the TnT T7 Insect Extract Protein Expression System (Promega, USA).Plasmid (4 μg) DNA template was added to the reaction mixture and the reaction was carried out at 29 °C for 4 h according to the manufacturer’s protocols. The protein productivity of the TnT T7 Insect Extract Protein kit is~50 μg/mL of the translation reaction mixture (Ezure et al.,2010; Golan-Mashiach et al., 2012).
Electrophoretic mobility shift assay (EMSA)
To verify differences in the DNA-protein binding capacity of MET1 to the putative E-box element sequence in theBmKr-h1promoter, EMSA was conducted using a LightShift Chemiluminescent EMSA Kit (Thermo Scientific, USA). The DNA oligonucleotides containing the consensus MET1 binding sites were labeled with biotin at the 5' end, incubated at 95 °C for 10 min, and annealed to generate the probe by natural cooling. Unbiotinylated probes were used as competitors to each other. The biotin-labeled oligonucleotides at the 5' end were synthesized by Invitrogen (China). The oligonucleotide probes used are shown in Supplementary Table S1.
The DNA-binding reactions were conducted in 20 μL of solution containing 150 ng of protein translatedin vitro,1×binding buffer, 50 ng of poly (dI-dC), 2.5% glycerol, 0.05%NP-40, 50 mmol/L KCl, 5 mmol/L MgCl2, 4 mmol/L EDTA, and 20 fmol of a biotinylated end-labeled double-stranded probe for 20 min at room temperature. For the competition assay,50- and 100-fold molar excesses of unbiotinylated doublestranded probe were added before adding the labeled probe.The gels were run at 100 V for 1.5 h using 6% polyacrylamide gel on ice. After electrophoresis, the gels were blotted onto positively charged Nylon membranes (Amersham Biosciences, USA). The membranes were then developed using the LightShift Chemiluminescent EMSA Kit according to the manufacturer’s protocol.
Construction of weighted gene co-expression networks
The FPKM values of DEGs in seven silkworm developmental stages (middle of last larval instar, end of the larval instar,wandering, pre-pupal, first day of pupal, middle of pupal,newly emerged moth) were calculated and used to construct a gene co-expression network using the WGCNA package in R v3.4.4 (Langfelder & Horvath, 2008). The gene co-expression modules were identified with the following parameters:networkType=“unsigned”, softPower=6 (B.mori) and 12 (B.mandarina), minModuleSize=30. The soft power value was chosen as a saturation level for a soft threshold of the correlation matrix based on the criterion of approximate scalefree topology. Briefly, genes with similar patterns of connection strengths to other genes or high topological overlap (TO) were selected and clustered based on their TO values to identify gene co-expression network groups. The gene co-expression network groups including theMet1gene were selected to draw the networks using Cytoscape v3.6.1(Li et al., 2017).
RESULTS
CRISPR/Cas9-mediated knockout of silkworm Met1 retarded brain development, in contrast to precocious metamorphosis of the cuticle
The genomic structure ofMet1was composed of one 1 545 bp long exon (Supplementary Figure S1A). After predicting the sgRNA sites (see Materials and Methods) followed byin vitroassay of activity of the corresponding sgRNAs(Supplementary Figure S1B), we selected one target site located at 563 bp for CRISPR/Cas9-mediated knockout.Consistent with previous research (Daimon et al., 2015), we found that 27.9% (56 of 201) of G0 larvae became severe larval-pupal mosaics during molting from the 3rd(L3) to 4th(L4)instar, with a brown-colored pupal cuticle that covered about half of the body (Figure 2A, B). Abdominal prolegs of some individuals showed degeneration (Supplementary Figure S2A). Most mosaic mutants failed to completely shed the old L3 cuticle (Supplementary Figure S2B). The homozygous mutations ofMet1were all lethal at the end of the L2 stage(Figure 2C). Given that the role of JH and its receptorMet1in the silkworm early larval stage remains elusive (Daimon et al.,2015) and that the mosaic mutants showed a clear phenotype of precocious metamorphosis of the cuticle, we used these mutants for further analysis.
Unlike the precocious metamorphosis in the cuticle, the mosaic mutant brain showed developmental retardation when compared with that of the WT (Figure 2D, E). The mosaic mutant brains retained the larval status and were significantly smaller (Figure 2D, E). In addition, PCA of the brain transcriptome data showed that the PC1 axis explained 63.5%of the variance in the samples, with younger to older samples distributed from right to left (i.e., L4 for WT, L5, wandering,and pupal stages), suggesting that this axis may be negatively associated with developmental status. The mosaic mutant brains were located on the right side of the WT L4 instar and differed from those of more mature developmental stages,e.g., L5 instar, wandering, and pupal stages (Figure 2F). This pattern suggests that the mosaic mutant brains remained at the earlier larval stage or suffered from developmental problems, rather than at the stage of precocious metamorphosis.
Genotyping of the mosaic mutant cuticles and brains showed that there were diverse types of indels in the genomic region spanning the target site, most of which caused frameshift mutations ofMet1(Figure 2G, H). The mutation rate was 46.7%-75.0% at the individual level (data not shown). These results confirmed that the somatic mosaic mutagenesis system was equally effective in both the cuticle and brain. In the silkworm brain,Met1mediated JH signaling functions in normal growth and likely promoted cell proliferation. Blockade of this pathway did not seem to activate precocious metamorphosis, as found inDrosophilabrains or other insect tissues (Abdou et al., 2011; Champlin &Truman, 1998;Daimon et al., 2015).
Kr-h1 was repressed in the epidermis and brain, whereas Br-C and E93 were overactivated in the epidermis in Met1 mutants
To study the molecular mechanisms underlying the differences in cuticle and brain responses toMet1depletion,we carried out comparative transcriptional analysis of the two tissues in L4 WT andMet1mosaics. A total of 5.98-12.12 Gb of clean data were generated for each sample and 73.30%-82.10% of the total clean reads were mapped to the silkworm genome (Supplementary Table S2). We investigated the expression levels of selected representative JH and 20E responsive genes. As expected, the expression ofKr-h1, a responsive gene of JH signaling, was significantly downregulated in the cuticle and brain ofMet1mutants (Figure 3A,B). These results confirmed that the JH pathway was blocked in both the cuticle and brain of the mutants. In the mutant cuticle, the pupal-specifier geneBr-C(Kayukawa et al., 2016)and adult-specifier geneE93(Kayukawa et al., 2017) were significantly up-regulated (Figure 3A).
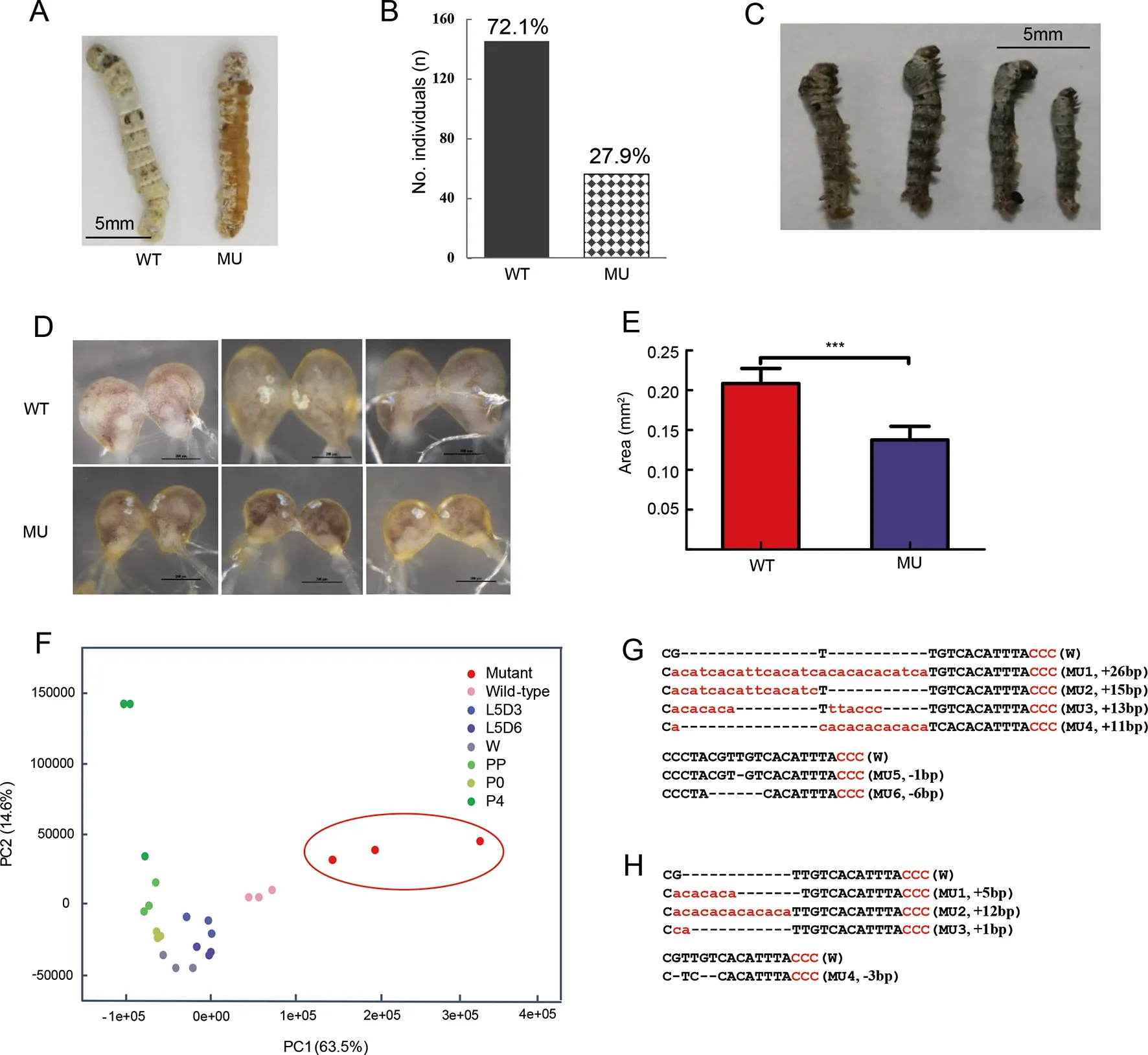
Figure 2 Phenotypes of CRISPR/Cas9-induced JH receptor mutants and Met1 mutation impaired brain development observed by cytological and transcriptome experiments
Epidermis and brain showed different transcriptomic pattern responses to Met1 depletion
We generated comparative transcriptomic analyses between the WT andMet1mosaics in both the epidermis and brain.There were 863 DEGs in the brain, nearly half the number found in the epidermis (1 523) (Figure 4A; Supplementary Tables S3, S4). A large proportion of DEGs (713, 82.6%) in the mutant brain were down-regulated (Figure 4A), possibly due to developmental arrest. In contrast, most DEGs were upregulated in the epidermis (1 101, 72.3%), suggesting general gene activation in precocious metamorphosis.
The two sets of DEGs were enriched in different pathways.In the epidermis, DEGs were mainly enriched in metabolic pathways involved in sugar metabolism and amino acid biosynthesis (Figure 4B). Up- and down-regulated genes were present in the metabolic processes, indicating active dynamics of metabolism in the mutant epidermis (Figure 4B). In addition,melanin synthesis genes, which account for the formation of the brown-colored pupal cuticle (Wang et al., 2017), were upregulated in the mutant epidermis (Supplementary Figure S3).These results indicate that the 20E-mediated pathways were triggered and functioned in the precocious pupation of the cuticle. Different from the active metabolic dynamics in the cuticle, enriched pathways in the brain were generally transcriptionally repressed, and included the highly enriched ECM-receptor interaction pathway, as well as the tyrosine metabolism, ABC transporter, retinol metabolism, and sugar metabolism pathways (Figure 4C). We compared the expression level of all genes in the ECM-receptor interaction pathway and found that they were markedly down-regulated in the mutant brain (Figure 4C). Protein digestion and absorption showed active dynamics, in contrast to active amino acid synthesis in the epidermis.
Artificial selection of Met1 potentially affected protein structure and weakly regulated the B. mori network
We recently identifiedMet1as a candidate domestication gene in the domestic silkwormB.mori(Xiang et al., 2018). We confirmed the selection signature ofMet1according to Xiang et al. (2018). The genomic region containingMet1demonstrated a strong selection signature with an elevatedFst andRODin theB.morigroup (Figure 5A).
Based on the above evidence showing thatMet1promotes silkworm larval brain development, and that the larval brain of domesticB.morihas fewer connections between the two lobes compared with wildB.mandarina(Figure 1), we further explored how artificial selection affected the biological function ofMet1during silkworm brain domestication.
We found strong differentiation in allelic frequency of theMet1gene bodies (Figure 5A). Three nonsynonymous substitution sites showed high divergence in allelic frequency between the domestic (B.mori) and wild (B.mandarina)silkworms (Figure 5A, B). Residue 104 in the basic helix-loop-

Figure 3 Gene expression gradient of representative genes of juvenile hormone (JH) and 20-hydroxyecdysone (20E) signaling pathways
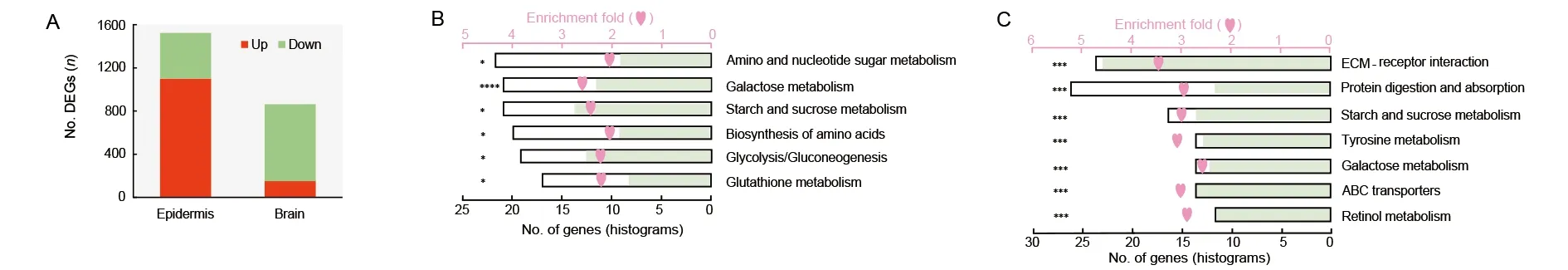
Figure 4 Illustration of comparative transcriptomics between Met1 mosaics and wild-type (WT)
helix (bHLH) transcriptional activity domain and residue 210 in the region linking the two PAS domains (Charles et al., 2011)appeared to be under positive selection in theB.moriclade when compared with other Lepidoptera species, as detected by PAML analyses (Table 1). In theB. morigroup, isoleucine and asparagine were relatively fixed at residues 104 and 210,respectively (Figure 5A, B), whereas all other Lepidoptera species carried valine and histidine, respectively (Figure 5B).TheB.moriMET1 showed different 3D structure and protein dynamical properties in the region containing residue 210,where the imidazole-cycle bearing histidine conserved in other Lepidoptera species was replaced by aliphatic asparagine in theB.moriaccording to prediction (Figure 5C-E). These results suggest that mutations located in the linker region of the two PAS domains influenced the structure of MET1.
To further test the functional impact of the MET1 mutations,we conducted EMSA within vitro-expressed MET1 proteins and the probe of the core binding region of its target geneKrh1(Kayukawa et al., 2012). We first tested the functional contribution of the linker region to MET1. Truncated MET1 without the linker region showed severely reduced binding activity with the labeled probes (Figure 6A), suggesting a key role of this region in transcriptional binding activity. We then compared the transcriptional binding activity of MET1 betweenB.moriandB.mandarina. Results indicated that MET1 had weaker binding activity with the labeled probe inB.morithan inB.mandarina(Figure 6A), suggesting that theB.moriallele in the linker region may contribute to the relatively weaker binding activity of MET1 as a transcription factor. In the competition assays, the specific band disappeared upon the addition of a 50- and 100-fold molar excess of unlabeled probe, indicating binding specificity.
To test whether the weaker binding activity ofB.moriMET1 affects the downstream regulatory network of the silkworm larval brain, we combined unpublished brain transcriptome data fromB.moriandB.mandarinato generate the weighted gene co-expression network module ofMet1in both species(see Materials and Methods) (Figure 6B, C). There were fewergenes co-expressed withMet1inB.mori(Figure 6B) than inB.mandarina(Figure 6C). Also, the connection weight between those genes andMet1was significantly lower inB.morithan inB.mandarina(Figure 6D). A weak co-expression network has also been reported in cultivated maize compared with its wild ancestor (Swanson-Wagner et al., 2012), implying that this mechanism may be common during rapidly evolved domestication. We suspect that the weak binding activity ofB.mori Met1may be associated with a weakened regulatory network in the brain during silkworm domestication. Whether and how this affects brain volume during silkworm domestication still needs further exploration.
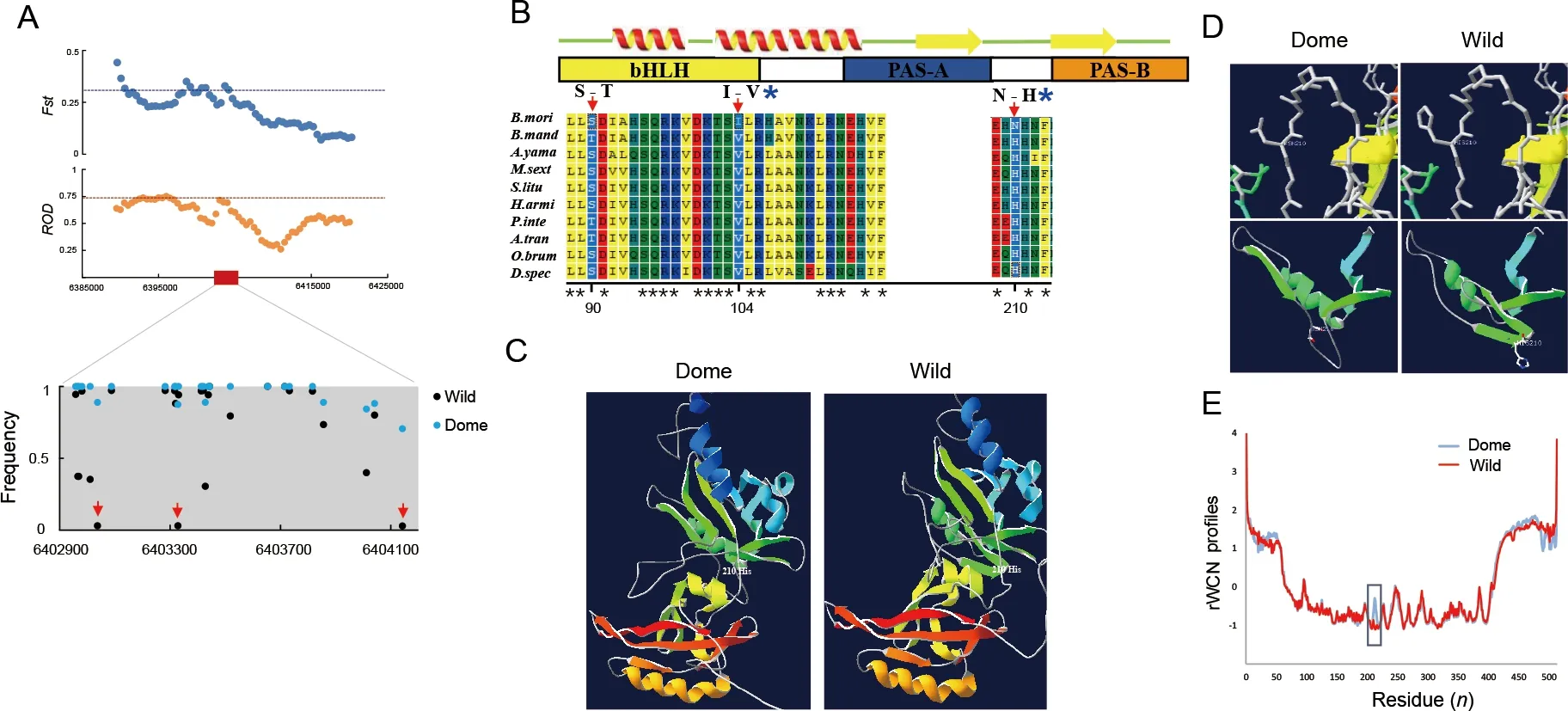
Figure 5 Selection of Met1 and analysis of mutations
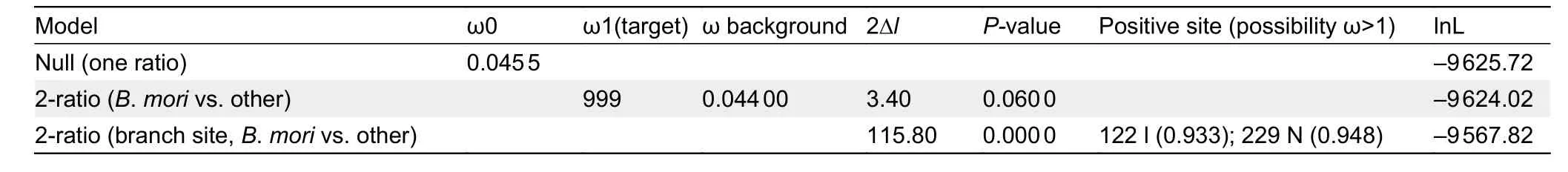
Table 1 PAML analysis of Met1 in Lepidoptera species

Figure 6 Influence of MET1 mutation on binding activity as a transcription factor and on co-expressed genes identified by weighted correlation network analysis (WGCNA)
Tyrosine metabolism pathway affected by JH signaling via Met1 may be involved in silkworm larval brain development and domestication
To further explore potentialMet1-affected transcriptomic changes in the larval brain during silkworm domestication, we performed comparative brain transcriptomic analyses betweenB.moriandB.mandarinaat three larval stages (Figure 1). We obtained 6.15-12.12 Gb of clean data for each sample(Supplementary Table S2). In total, 1 848, 3 953, and 2 962 DEGs were identified betweenB.moriandB.mandarinain the middle of the final larval stage, end of the final larval stage,and wandering stage, respectively (Supplementary Tables S5-S7). Almost no significantly enriched pathways were identified (Supplementary Table S8), but the tyrosine metabolism, ABC transporter, galactose metabolism, and ECM-receptor interaction pathways, which were enriched in the DEGs between the mutant vs. WT comparison(Figure 4C), were among the top ranked (but non-significant)enrichment results (Supplementary Table S8). We further overlapped two sets of DEGs, i.e., mutant vs. WT andB.morivs.B.mandarina, at the three larval stages, respectively, and performed KEGG enrichment analyses of the shared DEGs for each group. Notably, tyrosine metabolism was the common enriched pathway at the three larval stages (Figure 7A),suggesting that this pathway is involved in both larval brain development and domestication.
We investigated two core pathways in the tyrosine metabolism network (Figure 7B). One pathway involved the catalysis of tyrosine by peroxidase (POD) and peroxidasin(PXDN) to form tyrosine derivatives (Figure 7B). These two enzymes can generate tyrosine-tyrosine bonds to stabilize the ECM (Fessler et al., 1994). In the brain ofMet1mosaics, the DEGs encoding the two enzymes were all down-regulated(Figure 7B; Supplementary Figure S4), suggesting that depletion of JH signaling influenced the ECM of the brain(Figure 4C). Genes encodingPodin theB.moribrain were down-regulated compared with that inB.mandarina(Figure 7B; Supplementary Figure S4). However, when we compared the expression of all genes involved in the ECMreceptor pathway, we found no differential expression in the brain betweenB.moriandB.mandarina(Figure 7C).
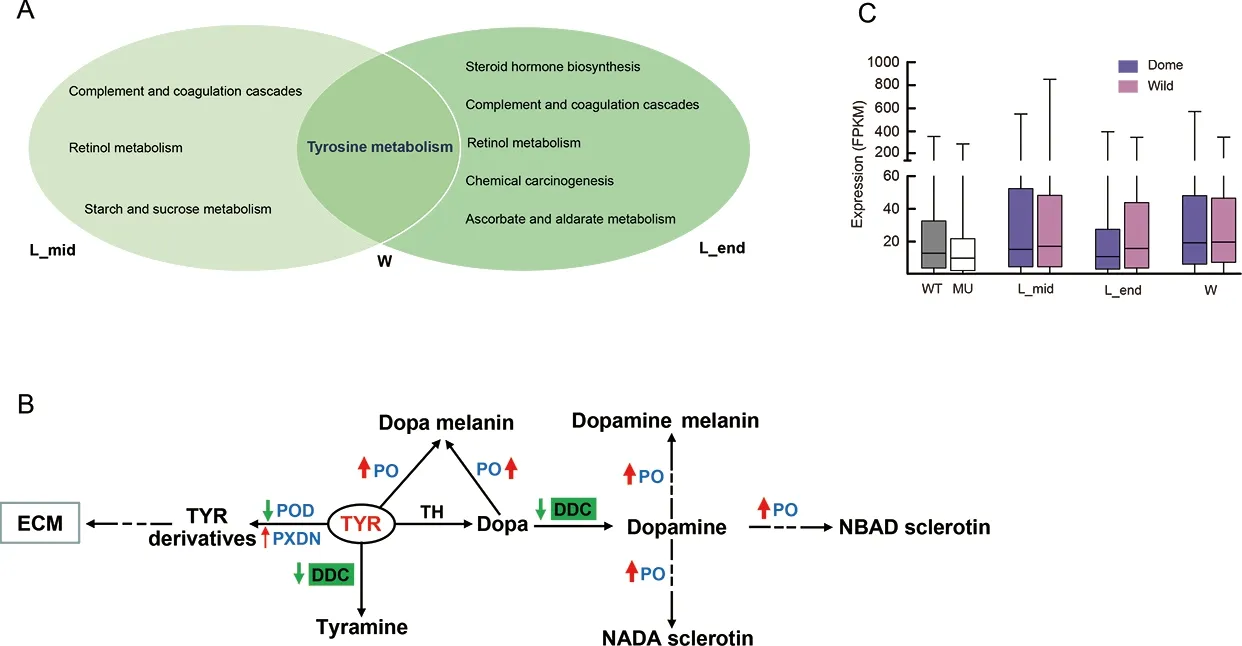
Figure 7 Illustration of comparative transcriptomics between B. mori and B. mandarina
The other pathway is the tyrosine-tyramine and dopaminepigmentation melanin synthesis pathway (Figure 7B). The three shared DEGs encode phenoloxidases (POs), which are at the distal end in melanin synthesis (Wang et al., 2017).They are involved in insect immunity and are considered important enzymes in the insect developmental process. For example, in the oriental fruit fly, RNAi knockdown of PO at the end larval stage impedes larval-pupal transition (Bai et al.,2014). Here, genes encoding PO in theMet1mutant brain were also down-regulated (Figure 7B), suggesting roles in brain development. Interestingly, genes encoding PO were upregulated in theB.moribrain compared to theB.mandarinabrain (Figure 7B) and DOPA decarboxylases (DDCs) were specifically down-regulated inB.mori(Figure 7B;Supplementary Figure S4). DDCs use tyrosine and L-dopa as substrates to synthesize tyramine and dopamine, respectively.Up-regulated POs and down-regulated DDCs may have resulted in a deficiency of tyramine and/or dopamine inB.moricompared withB.mandarina.
DISCUSSION
By generating mosaic loss-of-function mutants of the JH receptorMet1, we demonstrated that JH signaling promotes the development of the silkworm larval brain. However, this promotion could not be simply explained by repression of precocious metamorphosis, which is easily detected in the epidermis (Daimon et al., 2015; Zhu et al., 2019). Firstly, we found that theMet1mutant epidermis showed active dynamics of sugar metabolism and amino acid biosynthesis, which usually occur in tissues experiencing metamorphosis, whereas gene expression was largely repressed in the brain. Secondly,expression of the JH-responsive geneKr-h1was downregulated in both the epidermis and brain, whereas the genes related to metamorphosis (i.e.,Br-CandE93) were only upregulated in the epidermis, thus confirming the different responses of the two tissues toMet1defects. Further exploration of JH andKr-h1should improve our understanding of the regulation role of JH signaling in silkworm brain development. Thirdly, we identified several downstream pathways thatMet1may affect in the brain, including the ECM-receptor interaction and tyrosine metabolism pathways.The ECM consists of many families of molecules, including collagens, non-collagenous glycoproteins, glycosaminoglycans, and proteoglycans, and plays an important role in processes underlying the development, maintenance, and regeneration of the nervous system (Broadie et al., 2011).Here, down-regulation of this pathway in the silkworm mutant brain resulted in developmental arrest of the larval brain. In regard to the tyrosine metabolism pathway, which was generally repressed in theMet1mutants, we noted that a branch in this pathway, i.e., catalysis of tyrosine by POD and PXDN to form tyrosine derivatives, is related to the biosynthesis of thyroxine and its derivatives (Wang et al.,2017) (Supplementary Figure S5). In mammals, thyroxine is a very important hormone secreted by the thyroid and coordinates with growth hormone to regulate brain development (Roger & Fellows, 1979). While earlier studies have suggested that derivatives of thyroxine can mimic the effects of JH to influence egg production in some insects (Kim et al., 1999), whether this biosynthesis pathway is active in insects remains unknown, raising the question of the role of this hormone in the insect brain. On the other hand, the POD and PXDN enzymes can generate tyrosine-tyrosine bonds,which stabilize the ECM (Fessler et al., 1994), suggesting that the ECM-receptor interaction and tyrosine metabolism pathways may interact.
Based on molecular evolution, functional assay of potential causal replacement inMet1, and comparative brain transcriptome analyses betweenB.moriandB.mandarina,we explored the possible mechanism and biological impact of artificial selection on this domestication gene. We identified a novel motif of MET1 that may influence its function as a transcription factor and detected a notable amino acid replacement in this motif fixed inB.moriduring the domestication process. The amino acid replacement was predicted to affect the 3D protein structure and dynamic characteristics. Intriguingly, despite large-scale evolution in Lepidoptera, this replacement was still only specific in the domestic species, with a strong positive selection signature.All other tested wild species uniformly showed the other genotype at this site. We suspect that this artificially selected replacement may have had substantial biological impact on the adaptation of wild silkworms to the domestic environment.In domestic dogs, Axelsson et al. (2013) also identified candidate mutations in key genes of the starch digestion pathway, which showed higher catalytic activity, with some of these mutations also shared in herbivores. Our results suggested that theMet1mutation inB.morimay result in weaker binding activity to thecis-element of the JH-responsive geneKr-h1, and further influence the regulatory network.Evidence from crops suggests that modifications in transcription factors play important roles in domestication and affect agronomic traits in a pleiotropic manner (Gross & Olsen,2010). Here, the biological impact of the artificial selection ofMet1also influenced multiple aspects of silkworm domestication. JH-Met1-Kr-h1signaling functions in multiple tissues and shows complex crosstalk with other signaling pathways, and varies among tissues and species (Li et al.,2019; Riddiford, 2020; Shpigler et al., 2020). In the brain, we detected a weaker co-expression network ofMet1inB.morithan inB.mandarina. Although this predicted network requires further verification, it provides a preliminary landscape of the impact ofMet1artificial selection on brain gene expression patterns, which may further influence the development of the brain itself and the behaviors that the brain controls.
In the larval brain, comprehensive transcriptomic analyses indicated that tyrosine metabolism was a potential downstream pathway influenced by artificial selection of JH signaling. Its intermediate products, i.e., tyramine and dopamine, are important brain neurotransmitters and affect invertebrate behaviors such as locomotion, nutritional state,escape response, and flight (Riemensperger et al., 2011;Schützler et al., 2019; Vierk et al., 2009). For instance, inDrosophila, deficiency of dopamine results in reduced activity,extended sleep time, locomotor deficits, and hyperphagia,similar to that found inB.moricompared toB. mandarina.Previous research has reported that JH regulates these brain biogenic amine systems (Zhu et al., 2009). Therefore, we propose that artificial selection of JH signaling may have acted on tyrosine metabolism, thereby influencing the brain biogenic amine system, at least for larval behavioral changes, during silkworm domestication.
DATA AVAILABILITY
RNA-seq data were deposited in the NCBI under BioProjectID PRJNA756861 and were deposited in the Genome Sequence Archive (GSA) database under Accession No. CRA004783.
SUPPLEMENTARY DATA
Supplementary data to this article can be found online.
COMPETING INTERESTS
The authors declare that they have no competing interests.
AUTHORS’ CONTRIBUTIONS
Y.C. performed the experiments, collected and analyzed the data, and wrote the paper; Z.L.L. and L.Y. performed the microinjections; Y.J.L. performed resource collection; C.C.L.,Z.D.Z., X.M.W., and H.M.D. participated in data analysis; H.X.,Y.P.H., and Q.L.F. designed the experiment, analyzed the data, and wrote the manuscript. All authors read and approved the final version of the manuscript.
ACKNOWLEDGEMENTS
We thank Dr. An-Jiang Tan for the Cas9 gene template used in this work, Dr. Shuai Zhan for help in genomic data analyses, Yu-Ling Peng for help with microinjections, and Dr Mu-Wang Li for help in silkworm maintenance. We thank LetPub (www.letpub.com) for linguistic assistance during the preparation of this manuscript.
杂志排行

Zoological Research的其它文章
- Chromosome-scale genome assembly of brownspotted flathead Platycephalus sp.1 provides insights into demersal adaptation in flathead fish
- Global view on virus infection in non-human primates and implications for public health and wildlifeconservation
- Captopril alleviates lung inflammation in SARS-CoV-2-infected hypertensive mice
- Deletion of phosphatidylserine flippase β-subunit Tmem30a in satellite cells leads to delayed skeletal muscle regeneration
- Molecular phylogeny and morphological comparisons of the genus Hebius Thompson, 1913 (Reptilia:Squamata: Colubridae) uncover a new taxon from Yunnan Province, China, and support revalidation of Hebius septemlineatus (Schmidt, 1925)
- Prosecution records reveal pangolin trading networks in China, 2014-2019